NPLATE® Polvo para solución inyectable 250 mcg
TECNOFARMA
Otros hemostáticos sistémicos.
Composición.
Cada frasco ampolla contiene: *Romiplostim 375 mcg, manitol, sacarosa, L-histidina, ácido clorhídrico diluido y polisorbato 20 (*Para el vial de uso Unico de 250 mcg (conteniendo 375 mcg de romiplostim), al diluir con 0,72 ml agua esteril para inyectables, entrega 250 mcg/0,5 mL.
Indicaciones.
Nplate está indicado para el tratamiento de trombocitopenia en pacientes adultos con púrpura trombocitopénica (idiopática) (PTI) inmune crónica: Que no fueron sometidos a una esplenectomía y tuvieron una respuesta inadecuada o son intolerantes a los corticoides y/o a las inmunoglobulinas; Que fueron sometidos a una esplenectomía y tuvieron una respuesta inadecuada a la esplenectomía.
Dosificación.
El tratamiento debería ser supervisado por un profesional de la salud experimentado. Régimen de Dosis Recomendado: Nplate se administra en forma semanal como una inyección subcutánea con ajustes de dosis basados en la respuesta del recuento plaquetario. Utilice la dosis más baja de Nplate necesaria para lograr y mantener un recuento de plaquetas ≥ 50 x 109/L. La dosis de Nplate prescripta puede consistir en un volumen muy pequeño (por ejemplo, 0,15 mL). Nplate debería administrarse sólo con una jeringa con graduaciones de 0,01 mL. Dosis Inicial: La dosis inicial de Nplate es 1 mcg/kg basada en el peso corporal real. Ajustes de Dosis: Ajustar la dosis semanal de Nplate en incrementos de 1 mcg/kg hasta que el paciente logre un recuento plaquetario ≥ 50 x 109/L, pero ≤ 150 x 109/L. Evaluar el recuento plaquetario en forma semanal hasta que se logre un recuento plaquetario estable (≥ 50 x 109/L durante por lo menos 4 semanas sin ajuste de dosis). Después, obtener recuentos plaquetarios mensuales. No exceder la dosis máxima semanal de 10 mcg/kg. Ajustar la dosis como se muestra en la Tabla 3.
Debido a la respuesta interindividual plaquetaria variable, en algunos pacientes el conteo plaquetario puede caer repentinamente por debajo de 50 x 109/L después de la reducción de dosis o de la discontinuación del tratamiento. En estos casos, si es clínicamente apropiado, el nivel más alto de estratificación de conteo plaquetario para la reducción de la dosis (200 x 109/L) y la interrupción de tratamiento (400 x 109/L) puede considerarse de acuerdo al criterio del médico. Discontinuación del Tratamiento: Los pacientes deberían evaluarse clínica y periódicamente y el médico debería decidir la continuación del tratamiento teniendo en cuenta a cada paciente en particular. Si el recuento de plaquetas no se incrementa a un nivel suficiente, después de 4 semanas de administración de la dosis más alta de 10 mcg/kg, en forma semanal, discontinuar Nplate para evitar un sangrado clínicamente importante. Es probable que la trombocitopenia reaparezca tras la discontinuación del tratamiento. Uso de Nplate con Terapias Farmacológicas Concomitantes para la PTI: Las terapias farmacológicas para la PTI utilizadas en combinación con Nplate en estudios clínicos incluyeron corticoides, danazol y/o azatioprina, inmunoglobulina normal (IVIG) e inmunoglobulina anti-D Rho. Si el recuento plaquetario del paciente es > 50 x 109/L, otras terapias farmacológicas para la PTI pueden reducirse o discontinuarse. Reconstitución: Reconstituir sólo con Agua Estéril para Inyecciones como se detalla en la Tabla 4. No utilizar agua salina o bacteriostática para inyección al reconstituir el producto.
Un sobrellenado adicional se incluye en cada vial para asegurar que 250 mcg o 500 mcg de romiplostim pueda ser administrado (Tabla 4). Dado que el volumen de inyección puede ser muy pequeño, debería utilizarse una jeringa con graduaciones de 0,01 mL. Revolver el frasco ampolla ligeramente e invertirlo para reconstituir. No sacudir o agitar el frasco ampolla fuertemente. Generalmente, la disolución de Nplate lleva menos de 2 minutos. La solución reconstituida debe ser transparente e incolora. Antes de la administración, los productos parenterales deben inspeccionarse visualmente a fin de identificar material particulado y decoloración. Si se observan partículas o decoloración, no debe utilizarse el contenido del recipiente. Nplate debe utilizarse dentro de las 24 horas de la reconstitución. El producto reconstituido puede mantenerse en la jeringa por un máximo de 4 horas, aunque se recomienda que el producto reconstituido se utilice de inmediato después de la reconstitución. La esterilidad del producto, post-reconstitución, depende de las técnicas asépticas y del ambiente en que la dosis fue preparada. Durante el tiempo en que la reconstitución de romiplostim se mantenga en la jeringa, la temperatura no debe exceder los 25°C y debe ser protegido de la luz. El producto es para utilizarse una sola vez en un solo paciente. Desechar cualquier residuo. No deben agregarse otros medicamentos a las soluciones que contienen Nplate. Cálculo de la Dosis: Para determinar el volumen de inyección que deberá administrarse, primero identificar la dosis total de los pacientes en microgramos, empleando la información sobre dosificación en Dosificación (posología): Dosis Inicial y Ajustes de Dosis. Para calcular la dosis de Nplate, siempre debe utilizarse el peso corporal real al inicio del tratamiento. Por ejemplo, un paciente de 75 kg que inicia una terapia a 1 mcg/kg comenzará con una dosis de 75 mcg. El volumen de la solución de Nplate que deberá administrarse se calcula dividiendo la dosis en microgramos por la concentración de la solución de Nplate reconstituida (500 mcg/mL). En el ejemplo de este paciente, la dosis de 75 mcg se divide por 500 mcg/mL, lo cual da como resultado un volumen de inyección de 0,15 mL. Precauciones Durante la Administración: Se deberá tener precaución durante la preparación de Nplate en el cálculo de la dosis y la reconstitución con el volumen correcto de Agua Estéril para Inyectable. Se debe tener especial cuidado para asegurar que se retira el volumen adecuado de Nplate del frasco ampolla para la administración subcutánea. Precauciones en el uso: Pacientes con Insuficiencia Hepática o Renal: La experiencia es limitada en pacientes con insuficiencia hepática o renal severa. En estas poblaciones, Nplate debería usarse con precaución. Efectos sobre la Fertilidad: Romiplostim no tuvo un efecto observado sobre la fertilidad de ratas macho y hembra en dosis subcutáneas de hasta 100 mcg/kg administradas 3 veces por semana (hasta 9 veces el ABC en humanos en la dosis clínica máxima recomendada). No obstante, el valor predictivo de este estudio efectuado en animales es limitado debido al desarrollo frecuente de anticuerpos neutralizantes contra el medicamento. Uso en el Embarazo: La seguridad y eficacia de romiplostim en las mujeres embarazadas no se ha establecido. En estudios de toxicidad de desarrollo en ratas y conejos, no se observó evidencia de daño fetal por exposición de romiplostim de hasta 11 veces (ratas) y 82 veces (conejos) mayor que la dosis máxima indicada para humanos de 10 mcg/kg (ver sección Dosificación (posología)). En ratones, a exposiciones 5 veces mayor a la dosis máxima indicada para humanos, hubo reducciones en el peso corporal maternal y evidencia de incremento de pérdida postimplantación. En un estudio de desarrollo pre-natal y post-natal realizado en ratas, a exposiciones 11 veces a la dosis máxima en humanos, hubo un pequeño incremento en la incidencia de mortalidad perinatal de las crías. Romiplostim atraviesa la placenta en ratas a dosis clínicamente relevantes o mayores. Nplate no debería utilizarse durante el embarazo a menos que el posible beneficio justifique el posible riesgo al feto. Uso Durante el Período de Lactancia: Se desconoce si romiplostim se presenta en la leche humana. Muchos medicamentos se presentan en la leche humana, y por el potencial de efectos adversos de romiplostim en lactantes, debe tomarse una decisión de suspender la lactancia o suspender el medicamento, teniendo en cuenta el posible beneficio del medicamento para la madre o el beneficio posible de la lactancia para el lactante. Uso en Pacientes Pediátricos: No se determinaron la seguridad y la eficacia de Nplate en pacientes pediátricos ( < 18 años). Uso en Pacientes de Edad Avanzada: De los 204 pacientes que recibieron Nplate en estudios clínicos sobre PTI, 38 (19%) eran ≥ 65 años y 18 (9%) eran ≥ 75. No se observaron diferencias globales de seguridad o eficacia entre los pacientes de edad avanzada y los pacientes más jóvenes en los estudios controlados con placebo. Carcinogénesis: No se ha investigado el potencial carcinogénico de romiplostim. Hay una preocupación teórica de que romiplostim puede estimular la proliferación de células cancerosas existentes que expresan el receptor de la TPO (ver Advertencias: Progresión de Síndromes Mielodisplásicos (SMD) Existentes). Genotoxicidad: No se ha investigado el potencial genotóxico de romiplostim.
Contraindicaciones.
Nplate está contraindicado en pacientes con hipersensibilidad conocida a productos derivados de E. coli, romiplostim o cualquier otro componente del producto.
Reacciones adversas.
En la Tabla 1, se muestran los eventos adversos informados en ensayos clínicos en fase 3. Teniendo en cuenta el análisis de todos los pacientes que recibieron Nplate en dos estudios controlados con placebo, se informaron eventos adversos en 39 (95%) pacientes que recibieron placebo (n = 41) y en 84 (100%) que recibieron Nplate (n = 84). La mayoría de estos eventos fueron leves a moderados, aproximadamente el 27% se clasificaron como severos. Los eventos adversos que se informaron con más frecuencia fueron dolor de cabeza, artralgia y mareos. Usualmente, los dolores de cabeza fueron leves o moderados y se manejaron con analgésicos no narcóticos.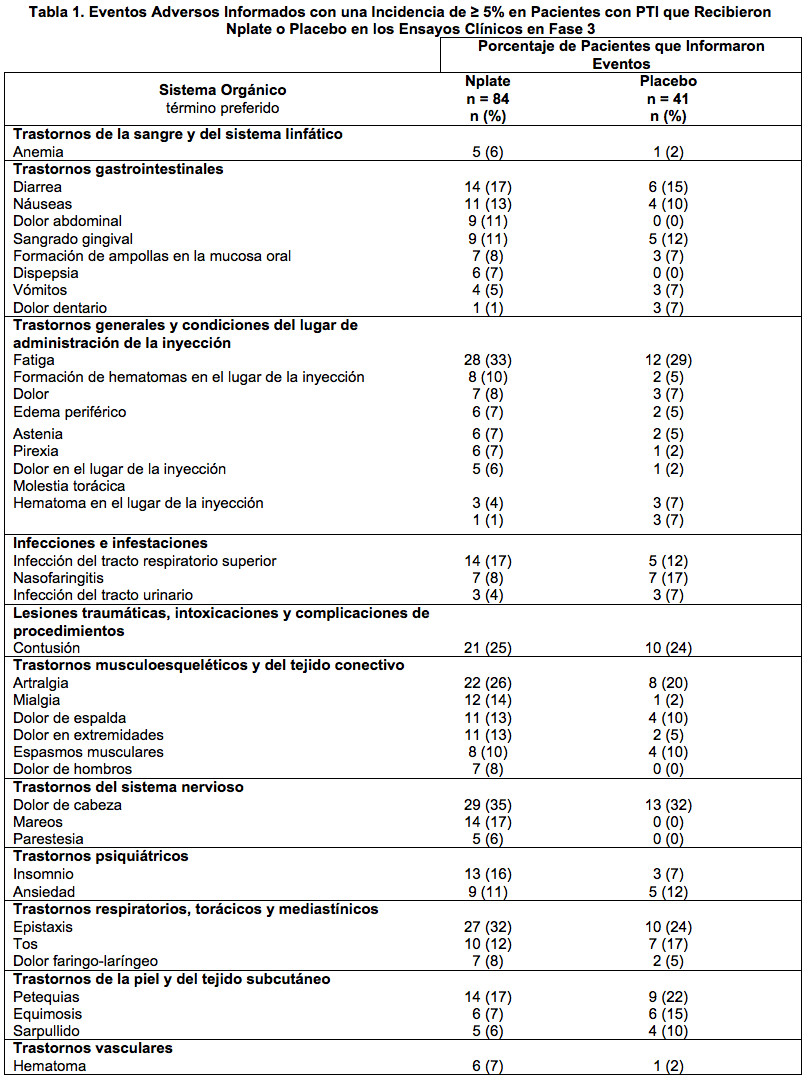
Eventos Adversos Serios/Muertes/Suspensiones/Intervenciones: Catorce pacientes (17%) tratados con Nplate experimentaron eventos adversos serios, dos (2%) que tuvieron 3 eventos adversos serios evaluados por el investigador como posiblemente relacionados con el tratamiento: trastorno de la médula ósea determinado como el aumento de reticulina, embolia periférica e isquemia periférica. Ocho (20%) pacientes tratados con placebo experimentaron eventos adversos serios. Hubo cuatro eventos adversos fatales durante los dos estudios controlados con placebo (1 paciente recibió Nplate (1%) y 3 pacientes recibieron placebo (7%)); ninguna de las muertes se asoció con el tratamiento. El paciente tratado con Nplate murió tras una hemorragia intracraneal que se produjo después que se interrumpió Nplate, ante la presencia de terapia antiplaquetaria. Los eventos adversos fatales en los pacientes tratados con placebo fueron (n (%)): hemorragia cerebral (1 (2%)), embolia pulmonar (1 (2%)) y neumonía atípica primaria tras la hospitalización durante un sangrado intracraneal (1 (2%)). Veinticinco pacientes interrumpieron el tratamiento: 5 (6,0%) pacientes que recibían Nplate y 20 pacientes (48,8%) tratados con placebo. Tres pacientes tratados con Nplate discontinuaron el tratamiento debido a eventos adversos serios: linfoma de células B en un paciente con linfadenopatía preexistente y diversos agregados linfoides en la médula ósea, trastorno de la médula ósea determinado como el aumento de reticulina y hemorragia intracraneal tras la discontinuación de Nplate mientras se administraba también terapia antiplaquetaria. Un paciente tratado con placebo discontinuó el estudio por metástasis en el hígado. Ochenta y tres por ciento de los pacientes en los grupos Nplate y placebo presentaron eventos adversos que llevaron a la intervención (por ejemplo, alteración o discontinuación de la medicación de estudio, otros medicamentos o terapias administradas, hospitalización). Los eventos adversos más frecuentes que condujeron a la intervención en los grupos Nplate y placebo, respectivamente, fueron dolor de cabeza (29% vs. 27%), infección del tracto respiratorio superior (13% vs. 10%) y artralgia (12% vs. 7%). Seguridad a Largo Plazo: El grupo de seguridad a largo plazo de fase 3 en relación con la PTI comprende a todos los pacientes que recibieron el producto investigado en los dos estudios sobre PTI en fase 3 más los datos de exposición y seguridad del estudio de ampliación a largo plazo. En el estudio de ampliación a largo plazo, se inscribieron un total de 117 pacientes de los dos estudios controlados con placebo: 83 habían recibido Nplate y 34 pacientes habían recibido placebo en los estudios controlados con placebo. La mediana de duración del tratamiento en la ampliación a largo plazo fue 39 semanas (rango: 5 a 84 semanas), con una mediana de dosis semanal de 3 mcg/kg. Se calcularon las tasas ajustadas por duración del estudio a fin de explicar las cantidades variables de tiempo en que los pacientes individuales se inscribieron en el estudio. Las tasas de incidencia de eventos adversos ajustadas por duración del estudio se expresaron como la cantidad de eventos cada 100 paciente-años en estudio. Ciento trece pacientes informaron 1731 eventos adversos mientras recibían Nplate respecto de una tasa de eventos ajustada por duración de 1782 eventos cada 100 paciente-años en estudio. Cuarenta pacientes informaron un total de 406 eventos adversos mientras recibían placebo respecto de una tasa de eventos ajustada por duración de 2143 eventos cada 100 paciente-años en estudio. Los eventos adversos más frecuentes en ambos grupos de tratamiento (tasas de eventos ajustadas por duración del estudio) con Nplate y placebo, respectivamente, fueron dolor de cabeza (131, 169), contusión (80, 127), epistaxis (80, 85) y fatiga (67, 106). Las tasas más altas de eventos adversos serios ajustadas por duración del estudio correspondieron a la disminución en el recuento de plaquetas (Nplate 15 vs. placebo 42). Análisis de Eventos de Sangrado Informados: En todos los ensayos clínicos sobre PTI, se observó una relación inversa entre los eventos de sangrado y los recuentos de plaquetas. Todos los eventos de sangrado clínicamente significativos (≥ grado 3) se produjeron en los recuentos de plaquetas < 30 x 109/L. Todos los eventos de sangrado > grado 2 se produjeron en los recuentos de plaquetas < 50 x 109/L. La incidencia de eventos de sangrado en estudios controlados con placebo figura en la Tabla 2. Nueve pacientes informaron un evento de sangrado que fue considerado serio (5 (6%) con Nplate, 4 (10%) con placebo). El 15% de los pacientes tratados con Nplate y el 34% de los pacientes tratados con placebo informaron eventos de sangrado grado 2 o más alto (ver Tabla 2).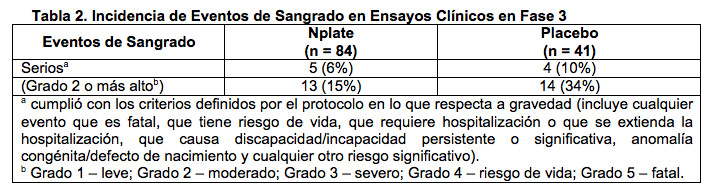
En cuanto al grupo de seguridad a largo plazo de fase 3 en relación con la PTI, la tasa de eventos ajustada por duración del estudio de los eventos de sangrado grado 2 o más alto fue 98 cada 100 paciente-años para los pacientes tratados con Nplate y 132 cada 100 paciente-años para los pacientes tratados con placebo. Estas tendencias en las tasas de eventos de sangrado se observaron en el contexto de una mayor reducción de medicación concomitante para la PTI entre pacientes que recibían Nplate en comparación con placebo. Además, hubo una mayor incidencia de uso de medicación de rescate entre pacientes que recibían placebo. Inmunogenicidad: Romiplostim no posee una secuencia de aminoácidos análoga a la trombopoyetina endógena (eTPO). Por lo tanto, es improbable que cualquier anticuerpo formado contra el producto presente reactividad cruzada con la eTPO. Como con todas las proteínas terapéuticas, hay un potencial de inmunogenicidad. Si sospecha la formación de anticuerpos neutralizantes, contactar a Tecnofarma S.A., teléfono: 2-5949201. En los estudios clínicos, la incidencia de anticuerpos preexistentes a romiplostim fue del 8% y la incidencia de desarrollo de anticuerpos de unión durante el tratamiento con Nplate fue del 6%. La incidencia de anticuerpos preexistentes a la TPO endógena fue del 5% y la incidencia del desarrollo de anticuerpos de unión a la TPO endógena durante el tratamiento con Nplate fue del 4%. De los pacientes con anticuerpos positivos a romiplostim o a la TPO, 2 (0,4%) pacientes tuvieron actividad neutralizante a romiplostim y ninguna tuvo actividad neutralizante a la TPO. Experiencia Poscomercialización: Se informaron casos de eritromelalgia. Se informaron casos de hipersensibilidad y angioedema.
Advertencias.
Las siguientes advertencias y precauciones especiales se han observado o son efectos teóricos de clase de estimuladores del receptor de la TPO. Recurrencia de Trombocitopenia Tras el Cese del Tratamiento: Tras la discontinuación de Nplate, la trombocitopenia puede reaparecer y, por ende, aumentar el riesgo de sangrado, sobre todo si Nplate se discontinúa cuando el paciente también está recibiendo anticoagulantes o agentes antiplaquetarios. Los pacientes deberían seguirse de cerca a fin de detectar una disminución en el recuento de plaquetas y ser controlados por el médico para evitar sangrado al discontinuar Nplate. Si se discontinúa el tratamiento con Nplate, se recomienda reiniciar la terapia para la PTI de acuerdo con las pautas de tratamiento actuales. El manejo médico adicional puede incluir el cese de la terapia con anticoagulantes y/o tratamiento antiplaquetario, la reversión de la anticoagulación o el soporte plaquetario. Aumento de la Reticulina en la Médula Ósea: Se ha observado reticulina en la médula ósea de algunos pacientes con PTI antes del tratamiento con Nplate y pareció aumentar en algunos pacientes tratados con Nplate. Se cree que la reticulina incrementada en médula ósea se debe al aumento del número de megacariocitos en la médula ósea, que posteriormente podrían liberar citocinas. En estudios clínicos con Nplate, la reticulina no se asoció con efectos clínicos adversos, casos de mielofibrosis idiopática crónica (MIC) o mielofibrosis secundaria y puede mejorar tras la discontinuación de Nplate. El aumento en la reticulina puede detectarse mediante una biopsia de la médula ósea y puede indicarse por cambios morfológicos en las células sanguíneas periféricas. Antes del y durante el tratamiento con Nplate, analice los frotis de sangre periférica y el conteo sanguíneo completo a fin de identificar nuevas anomalías morfológicas o anomalías morfológicas agravadas (por ejemplo, glóbulos rojos con forma de lágrima y nucleados, leucocitos inmaduros) o citopenia(s). Si un paciente desarrolla nuevas anomalías morfológicas o anomalías morfológicas agravadas o citopenia(s), interrumpir el tratamiento con Nplate y considerar efectuar una biopsia de médula ósea, con tinción adecuada para detectar fibrosis. También debería tenerse en cuenta el análisis citogenético de la muestra de médula ósea a fin de identificar una anomalía clonal. Complicaciones Trombóticas/Tromboembólicas: Los recuentos de plaquetas por encima del rango normal presentan un riesgo teórico de complicaciones trombóticas/tromboembólicas. Las incidencias de eventos trombóticos/tromboembólicos observadas en los grupos control fueron comparables a Nplate en estudios clínicos. No se observó una asociación entre estos eventos y los recuentos elevados de plaquetas. Se debe cumplir con las normativas respecto de los ajustes de dosis (ver sección Dosificación (posología)). En el contexto posterior a la comercialización, se han observado eventos trombóticos/tromboembólicos. Se debe tener precaución cuando se administre romiplostim a pacientes con factores de riesgo conocidos de tromboembolismo incluyendo, pero no limitados a, factores hereditarios (por ej. Factor V Leiden) o factores de riesgo adquiridos (por ej. deficiencia ATIII, síndrome antifosfolipídico), edad avanzada, pacientes con periodos prolongados de inmovilización, neoplasias, anticonceptivos y terapia hormonal sustitutiva, cirugía/traumatismo, obesidad y fumadores. Se han notificado casos de acontecimientos tromboembólicos (ATEs), incluyendo trombosis venosa portal, en pacientes con enfermedad hepática crónica que reciben romiplostim. Romiplostim debe utilizarse con precaución en estas poblaciones. Deben seguirse las recomendaciones para el ajuste de la dosis. Progresión de Síndromes Mielodisplásicos (SMD) Existentes: Los estimuladores del receptor de la TPO son factores de crecimiento hematopoyético que llevan a la expansión de la célula madre trombopoyética, a la diferenciación y la producción de plaquetas. El receptor de la TPO se expresa predominantemente sobre la superficie de las células de la línea mieloide. No hay expresión confirmada del receptor de la TPO sobre tumores sólidos. Con respecto a los estimuladores del receptor de la TPO, existe una preocupación teórica de que éstos pueden estimular la progresión de SMD existente. El diagnóstico de la PTI en adultos y en pacientes de edad avanzada se debería haber confirmado por la exclusión de otras entidades clínicas que presentan trombocitopenia, en particular se debe excluir el diagnóstico de SMD. En el caso de pacientes con síntomas sistémicos o signos anormales, por ejemplo blastocitos periféricos aumentados, normalmente se debería haber realizado aspirado y biopsia de médula ósea en el curso de la enfermedad y tratamiento, sobre todo en pacientes mayores de 60 años de edad. En estudios clínicos de tratamiento con Nplate en pacientes con SMD, hubo casos reportados de progresión a leucemia mieloide aguda (LMA), un potencial resultado clínico de SMD. Además, hubo casos de aumentos transitorios de blastocitos, que no progresaron a LMA. No se estableció el perfil de riesgo-beneficio para Nplate en SMD u otras poblaciones de pacientes sin púrpura trombocitopénica idiopática (PTI). Pérdida de Respuesta a Nplate: Una pérdida de respuesta o el hecho de no mantener una respuesta plaquetaria con Nplate debería impulsar la búsqueda de factores causales, incluso anticuerpos neutralizantes contra Nplate y un aumento de reticulina de la médula ósea. Errores de Medicación: Se han reportado errores de medicación, incluida la sobredosis y la administración insuficiente de dosis, en pacientes que reciben Nplate. La sobredosis podría generar un aumento excesivo en los recuentos plaquetarios asociado con complicaciones trombóticas/tromboembólicas. Si los recuentos plaquetarios aumentan excesivamente, discontinuar el tratamiento con Nplate y monitorear los recuentos plaquetarios. Reiniciar el tratamiento con Nplate de acuerdo con las recomendaciones de dosis y administración. Una administración insuficiente de dosis podría causar recuentos de plaquetas más bajos de lo esperado y posible sangrado. Los recuentos plaquetarios deben ser monitoreados en pacientes que reciben Nplate (ver secciones Dosificación (posología), Advertencias y síntomas y tratamiento de sobredosis). Efectos de Romiplostim sobre los Glóbulos Rojos y Blancos: Se han observado alteraciones en parámetros relacionados con los glóbulos rojos (disminución) y blancos (incremento) en ensayos toxicológicos no-clínicos (ratas y monos) pero no en pacientes con PTI. Debería considerarse la monitorización de dichos parámetros en los pacientes tratados con romiplostim.
Presentación.
Nplate está disponible en un envase que contiene 1 frasco de ampolla de: presentación de 250 mcg/0,5 mL: 375 mcg de romiplostim.


 Cambiar de país
Cambiar de país