XYNTHA®
PFIZER
Anticoagulante. Antihemofílico.
Composición.
Viales de único uso que nominalmente contienen 250, 500, 1000 o 2000 Unidades Internacionales (UI) de moroctocog alfa (factor VIII de la coagulación recombinante) por vial. Excipientes Polvo Liofilizado: Polisorbato 80, Sacarosa, L-Histidina, Cloruro de Calcio Dihidrato, Cloruro de Sodio, HCl. Excipientes diluyente: Cloruro de sodio y agua para inyectables.
Toxicología.
No se han realizado estudios con XYNTHA® para evaluar su potencial mutágeno o carcinógeno. XYNTHA® ha demostrado similitud con el producto predecesor ReFacto con relación a sus propiedades bioquímicas y fisicoquímicas, y en su farmacología y toxicología in vivo no clínica. Por inferencia se podría esperar que el producto predecesor ReFacto y XYNTHA® tengan potencial mutágeno y carcinógeno equivalentes. El producto predecesor ReFacto demostró en el ensayo de micronúcleos de ratón que no es genotóxico. No se han realizado estudios en animales para evaluar la carcinogenia, el deterioro de la fertilidad o del desarrollo fetal. En estudios preclínicos, XYNTHA® se utilizó para restauración segura y efectiva de la hemostasia. XYNTHA® demostró un perfil toxicológico similar al perfil toxicológico observado en el producto predecesor ReFacto, que a su vez ha demostrado perfil toxicológico similar a un producto del factor VIII derivado del plasma cuando se someten a pruebas de dosis repetidas en estudios toxicológicos con animales.
Indicaciones.
XYNTHA® está indicado para el control y prevención de episodios hemorrágicos y para la profilaxis de rutina y quirúrgica en pacientes con hemofilia A (deficiencia congénita del factor VIII o hemofilia clásica). XYNTHA® también está indicado para niños de todas las edades, incluyendo recién nacidos.
Dosificación.
Posología: El tratamiento con XYNTHA® debe iniciarse bajo supervisión de un médico con experiencia en el tratamiento de la hemofilia A. La posología y duración del tratamiento dependerá de la gravedad de la deficiencia del factor VIII, el lugar y nivel de hemorragia y la condición clínica del paciente. La respuesta al factor VIII puede variar en cada paciente, lográndose niveles diferentes de recuperación in vivo y demostrando diferentes vidas medias. Las dosis administradas se deben titular de acuerdo con la respuesta clínica del paciente. En presencia de un inhibidor, es posible que se necesiten dosis mayores o tratamientos específicos apropiados. No se ha evaluado en estudios clínicos el ajuste de la dosis en pacientes con insuficiencia renal o deterioro hepático. XYNTHA® puede utilizarse en adultos y niños. Ver también "Uso pediátrico". El número de unidades de factor VIII administradas se expresa en Unidades Internacionales (UI), que corresponden con la norma vigente de la OMS para productos del factor VIII. La actividad plasmática del factor VIII se expresa como un porcentaje (con relación al plasma humano normal) o en Unidades Internacionales (con relación al Estándar Internacional para el factor VIII en el plasma). Una Unidad Internacional (UI) de la actividad del factor VIII corresponde aproximadamente a la cantidad de factor VIII en un ml de plasma humano normal. El cálculo de la dosis requerida de factor VIII se basa en el hallazgo empírico de que, en promedio, una (1) UI de factor VIII por kg de peso corporal aumenta la actividad plasmática del factor VIII en 2 UI/dL. La dosis requerida se determina utilizando la siguiente fórmula: Unidades requeridas = peso corporal (kg) x aumento deseado del factor VIII (% del valor normal o UI/dL) x 0,5 (UI/kg por UI/dL). La potencia especificada en la etiqueta de XYNTHA® se basa en el ensayo de sustrato cromogénico de la Farmacopea Europea que se utiliza para calibrar el estándar de potencia en el proceso de fabricación de Wyeth utilizando un ensayo de coagulación de una fase. Este método de asignación de potencia está diseñado para armonizar XYNTHA® con el control clínico que utiliza un ensayo de coagulación de una fase. Con los productos del factor VIII recombinante, los controles clínicos que utilizan el ensayo cromogénico normalmente obtienen resultados que son mayores a los resultados obtenidos con el ensayo de coagulación de una fase. Los datos clínicos respaldan la utilización del ensayo de coagulación de una fase para controlar la terapia con XYNTHA®. Con base en su régimen de tratamiento actual, deberá recomendarse a las personas con hemofilia A que, cuando realicen un viaje, lleven el suministro adecuado del producto del factor VIII para los tratamientos necesarios. Deberá recomendarse a los pacientes consultar con su médico antes de viajar. Debe considerarse el control preciso de la terapia sustitutiva utilizando un ensayo de la actividad plasmática del factor VIII, particularmente durante intervención quirúrgica. Posología para Hemorragia y Cirugía: En caso de presentarse los siguientes eventos hemorrágicos, se debe mantener la actividad del factor VIII a los niveles plasmáticos o niveles superiores (en % del valor normal o en UI/dL) establecidos a continuación para el periodo de duración indicado.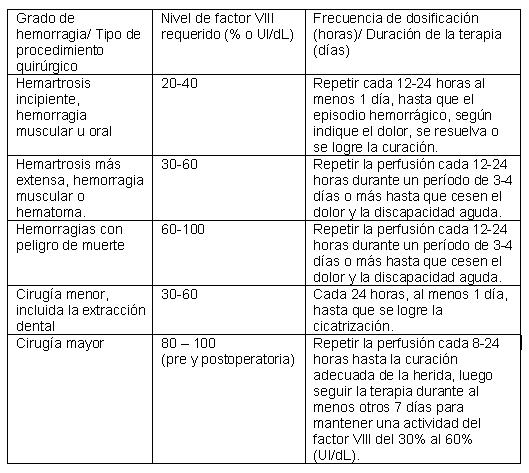
Durante el curso del tratamiento, se aconseja una determinación apropiada de los niveles del factor VIII para establecer la dosis a administrar y la frecuencia de las perfusiones repetidas. En el caso de intervenciones de cirugía mayor en particular, es indispensable la monitorización precisa de la terapia de sustitución por medio de análisis de la coagulación (actividad de factor VIII plasmático). La respuesta al factor VIII de cada paciente individual puede variar, consiguiendo diferentes niveles de recuperación in vitro y demostrando diferentes semividas. Posología para Profilaxis: En la profilaxis a largo plazo contra episodios hemorrágicos en pacientes con hemofilia A grave, las dosis habituales son de 20 a 40 UI de factor VIII por kg de peso corporal, en intervalos de 2 a 3 días. En algunos casos, especialmente en pacientes jóvenes, puede ser necesario un intervalo de dosificación más corto o dosis mayores. En los pacientes en tratamiento sustitutivo con factor VIII se debe controlar el desarrollo de inhibidores del factor VIII. Si no se obtienen los niveles de actividad plasmática de factor VIII esperados, o si el sangrado no se controla con la dosis adecuada, deben realizarse ensayos para determinar la presencia de inhibidores del factor VIII. Los datos de los ensayos clínicos indican que si están presentes inhibidores a niveles menores a 10 Unidades Bethesda (UB), la administración del factor antihemofílico adicional puede neutralizar los inhibidores. En pacientes con niveles de inhibidor superiores a 10 UB, la terapia con factor VIII puede no ser eficaz, y deben considerarse con otras opciones terapéuticas. El manejo de tales pacientes debe dirigirse por médicos con experiencia en el cuidado de pacientes con hemofilia. Inhibidores: Los pacientes que utilizan terapia sustitutiva del factor VIII se deben controlar para determinar si se desarrollan inhibidores del factor VIII. Si los niveles plasmáticos esperados de la actividad del factor VIII no se obtienen o si la hemorragia no se controla con la dosis apropiada, se debe realizar un ensayo para determinar la presencia de inhibidores del factor VIII. En pacientes con inhibidores (especialmente con niveles altos de inhibidores, mayores de 5 Unidades Bethesda, UB), el tratamiento con el factor VIII podría no ser efectivo y deberán considerarse otras opciones terapéuticas. El manejo de estos pacientes debe ser dirigido por médicos con experiencia en el cuidado de pacientes con hemofilia. Ver también "Precauciones" y Reacciones adversas". Administración: XYNTHA® Factor Antihemofílico (Recombinante), Libre de Plasma/Albúmina se administra por infusión intravenosa (IV) a lo largo de varios minutos después de la reconstitución del polvo liofilizado con la jeringa prellenada suministrada con diluyente (solución de Cloruro de Sodio 0,9%, 4 mL). Inspeccionar antes de la administración la solución y el contenedor para determinar la presencia de material particulado y decoloración. XYNTHA® Factor Antihemofílico (Recombinante), Libre de Plasma/Albúmina se debe administrar utilizando el juego de infusión suministrado en este kit y la jeringa prellenada de diluyente suministrada o una única jeringa plástica desechable estéril. Adicionalmente, la solución se debe extraer del vial utilizando el adaptador de vial. 1 Conecte la jeringa al extremo del equipo de infusión suministrado. 2. Coloque un torniquete y prepare el sitio de inyección limpiando bien la piel con una de las torundas de alcohol suministradas con el kit.
3. Realice la venopunción como se lo indicó su médico. Inserte la aguja del equipo de infusión dentro de la vena como se lo indicó su médico y retire el torniquete. Remueva el aire presente en el equipo de infusión retrayendo la jeringa. El producto XYNTHA® reconstituido se debe inyectar vía intravenosa durante varios minutos. La tasa de administración se debe determinar de acuerdo con el nivel de comodidad del paciente.
Luego de finalizar el tratamiento con XYNTHA®, retire el equipo de infusión y deséchelo. Descarte toda la solución no utilizada, los viales vacíos, las agujas y jeringas utilizadas depositándolos dentro de un recipiente apropiado para eliminación de desechos que pueden hacer daño a los demás si no se manipulan adecuadamente.
Contraindicaciones.
XYNTHA® puede estar contraindicado en pacientes con hipersensibilidad a alguno de los componentes de la preparación. XYNTHA® no se ha estudiado en pacientes con antecedentes de hipersensibilidad a las proteínas de hámster. XYNTHA® puede estar contraindicado en pacientes con antecedentes de hipersensibilidad a alguno de los componentes de la preparación y en pacientes con antecedentes de hipersensibilidad a proteínas de hámster.
Reacciones adversas.
En las siguientes tablas, se utilizan las categorías de frecuencia y los términos de CIOMS: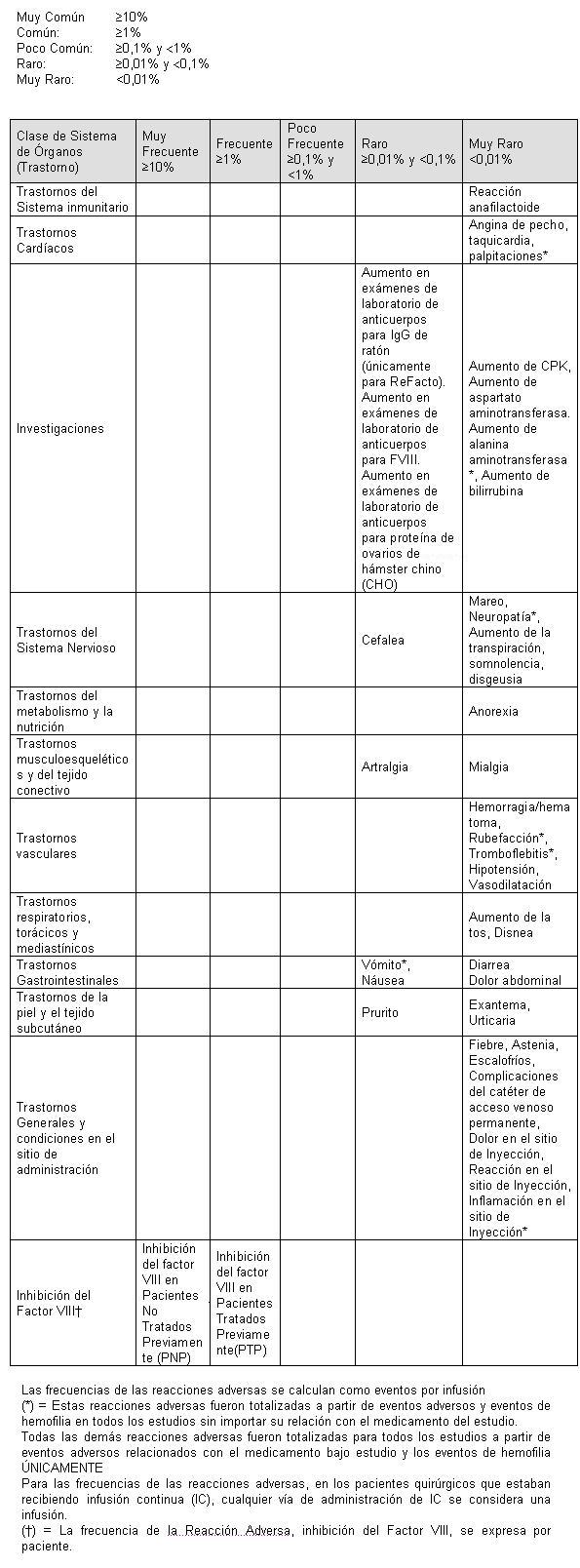
La reacción adversa debida al tratamiento por infusión, reportada con mayor frecuencia, fue el vómito. La mayoría de las reacciones reportadas fueron consideradas de severidad leve o moderada. Además, como ocurre con todos los productos intravenosos de proteínas, son posibles reacciones de hipersensibilidad del tipo alérgicas. Las manifestaciones de reacciones de hipersensibilidad pueden incluir urticaria, urticaria generalizada, presión en el pecho, sibilancias, hipotensión y anafilaxia. Si ocurre alguna reacción que se considera relacionada con la administración de Moroctocog Alfa (AF-CC), la tasa de infusión debe disminuirse o interrumpir la infusión, como lo indique la respuesta del paciente. Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizantes (inhibidores) para el Factor VIII. Ver también Inhibidores y Precauciones. Como ocurre con todos los productos del factor de coagulación VIII, debe controlarse a los pacientes con relación al desarrollo de inhibidores que son cualificados en Unidades Bethesda (UB) utilizando la modificación de Nijmegen del ensayo Bethesda. Si dichos inhibidores ocurren, la condición puede manifestarse por sí misma como una respuesta clínica insuficiente o un rendimiento bajo inesperado de la actividad plasmática del Factor VIII. En tales casos, se recomienda contactar un centro especializado en hemofilia. El riesgo de desarrollar inhibidores se correlaciona con la exposición al factor VIII antihemofílico, este riesgo es el más alto durante los primeros 20 días de exposición. Raramente, pueden desarrollarse inhibidores después de los primeros 100 días de exposición. Se han observado casos de inhibidor recurrente (titulación baja) después de cambiar desde un producto de FVIII a otro en pacientes previamente tratados con más de 100 días de exposición que tienen antecedentes previos de desarrollo de inhibidores. Por lo tanto, se recomienda controlar cuidadosamente a los pacientes con relación a la ocurrencia de inhibidores cuando se realice cambio a otro producto. Se han recibido informes de falta de efecto, principalmente en pacientes bajo profilaxis durante ensayos clínicos y en entornos postcomercialización. La falta de efecto y/o la baja recuperación del factor VIII se han reportado en pacientes con inhibidores pero también en pacientes que no presentan ninguna evidencia de inhibidores. La falta de efecto se ha descrito como hemorragia en las articulaciones objetivo, hemorragia en nuevas articulaciones, otras hemorragias o sensaciones subjetivas del paciente de inicio de nuevas hemorragias. Para garantizar una respuesta terapéutica adecuada es importante, Titular y controlar individualmente la dosis de XYNTHA de cada paciente, especialmente cuando se inicia el tratamiento con XYNTHA. En un estudio de fase 3 principal (estudio 310), en el que pacientes previamente tratados (PPT) con hemofilia A recibieron XYNTHA para profilaxis de rutina y tratamiento por demanda, 94 pacientes recibieron al menos una dosis de XYNTHA que resultó en un total de 6775 infusiones. En este estudio, la incidencia de los inhibidores del FVIII para XYNTHA fue el criterio de valoración de la seguridad principal. En dos pacientes con titulación baja, de estos 94 pacientes (2,1%), se observaron inhibidores transitorios. En un estudio complementario de XYNTHA (estudio 306), fueron observados 1 inhibidor de novo y dos inhibidores recurrentes (todos con titulación baja determinada por el laboratorio central) en 110 pacientes; la mediana de la exposición fue de 58 días de exposición (DE) (intervalo 5-140) y 98 pacientes tuvieron al menos 50 DE a XYNTHA. Noventa y ocho (98) de los 110 pacientes originales continuaron el tratamiento en un segundo estudio complementario (307) y extendieron la exposición a XYNTHA con una mediana de 169 DE adicionales (intervalo 9-425). Se observó un (1) inhibidor adicional de novo de baja titulación. La frecuencia de inhibidores observada en estos estudios se encuentra dentro del intervalo esperado. En un ensayo clínico de XYNTHA fabricado mediante el proceso anterior (estudio 300), uno de los 113 pacientes (0,9%) sometidos previamente a múltiples tratamientos que fueron evaluados para eficacia en episodios hemorrágicos desarrollaron inhibidor de alta titulación. El desarrollo de inhibidor en este paciente ocurrió en el mismo lapso de tiempo que el desarrollo de gammapatia monoclonal de importancia incierta. El paciente fue inicialmente observado en un laboratorio local presentando inhibidor de baja titulación debida al tratamiento a los 98 días de exposición lo que se confirmó a 2 UB en el laboratorio central a los 113 días de exposición. Después de 18 meses bajo tratamiento con XYNTHA, la concentración del inhibidor aumentó a casi 13 UB y un episodio de hemorragia no respondió al tratamiento con XYNTHA. En un análisis estadístico Bayesiano, los resultados del estudio 310 (dos de 94 pacientes desarrollaron un inhibidor, 89 presentaron 50 o más días de exposición a XYNTHA) se utilizaron para actualizar los resultados de los PPT de estudios complementarios previos de XYNTHA, en los que se observó un inhibidor de novo y dos inhibidores recurrentes en 110 pacientes y un inhibidor fue observado en 113 pacientes. Este análisis Bayesiano indica que la tasa de población con inhibidor (verdadero) para XYNTHA estuvo bajo un valor aceptable preestablecido del 4,4%; el estimado del límite superior del 95% de la tasa de inhibidor verdadero fue de 4,07%. En un estudio principal de fase 3 de profilaxis quirúrgica en pacientes con hemofilia A (estudio 311), un inhibidor persistente de baja titulación y un inhibidor falso-positivo transitorio fueron reportados. En un ensayo clínico (estudio 301), 32 de 101 pacientes (32%) previamente no tratados sometidos a tratamiento con XYNTHA [fabricado por el proceso anterior] desarrollaron inhibidores: 16 de 101 (16%) con una titulación > 5 Unidades Bethesda (UB) y 16 de 101 (16%) con una titulación ≤5 UB. La mediana de los días de exposición antes de desarrollar inhibidores en estos pacientes fue 12 días (intervalo 3 - 49 días). De los 16 pacientes que presentaron alta respuesta al tratamiento, 15 recibieron tratamiento de tolerancia inmunitaria (TI). Once (11) de los que presentaron respuesta alta al tratamiento tuvieron una titulación < 0,6 UB en su más reciente prueba disponible después del TI. Además, el tratamiento TI se inició en 10 de los 16 pacientes con baja titulación (≤5 UB), 9 de los cuales presentaron titulación < 0,6 UB para su valor más reciente. Por lo tanto, el TI mostró una eficacia general del 80% (20/25), 73% para los que presentaron alta respuesta el tratamiento y 90% para los que presentaron baja respuesta al tratamiento. En cinco (5) de los 6 pacientes restantes que presentaron baja respuesta y que no recibieron TI también se observó una titulación < 0,6 UB para su más reciente valor. Han existido informes postcomercialización espontáneos de desarrollo de inhibidores de alta titulación en pacientes previamente tratados. Se han observado en ensayos clínicos aumentos en el laboratorio de las titulaciones de anticuerpos anti-FVIII, sin desarrollo de inhibidores. En un estudio de PTP que estaban recibiendo XYNTHA para tratamiento de rutina y prevención de episodios hemorrágicos (estudio 310) y para profilaxis quirúrgica (estudio 311), 1 de 94 pacientes (1%), y 1 de 30 pacientes (3%), respectivamente, desarrollaron anticuerpos anti-FVIII; estos pacientes no desarrollaron un inhibidor. La significancia clínica de estos anticuerpos, en ausencia de un inhibidor no es clara. En ensayos clínicos de PTP que estaban recibiendo XYNTHA para tratamiento y prevención de rutina de episodios hemorrágicos, 0 de 94 pacientes (0%) en el estudio 310 y 3 de 110 pacientes (3%) en el estudio 306/307, desarrollaron un aumento en el laboratorio en la titulación de anticuerpos anti-CHO (ovarios de hámster chino, la estirpe celular que es la fuente del factor VIII para XYNTHA), sin ningún efecto clínico aparente. En un estudio con XYNTHA para profilaxis quirúrgica (estudio 311) 1 de 30 pacientes (3%) desarrolló un aumento en el laboratorio de anticuerpos para CHO. Veinte (20) de 113 (18%) PTP que estaban recibiendo XYNTHA fabricado mediante el proceso anterior (estudio 300) presentaron aumento en la titulación de anticuerpos anti-CHO, sin ningún efecto clínico aparente. La seguridad de XYNTHA fue evaluada en niños y adolescentes previamente tratados (n=18, edad 12-16 en un estudio [estudio 310]; y n=49, edad 7-16 en un estudio complementario [estudio 306]). Aunque se ha estudiado un número limitado de niños, existe una tendencia de mayores frecuencias de eventos adversos en niños de 7-16 años de edad cuando se comparan con los adultos. No existe ningún dato clínico de pacientes no tratados previamente (PNP) que se sometieron a tratamiento con XYNTHA. Los ensayos clínicos que evalúan la utilización de moroctocog alfa (AF-CC) en PNP (estudio 4434), en niños menores de 6 años de edad (estudio 313) y niños menores de 12 años de edad (estudio 4433) están en curso.
Advertencias.
Advertencias especiales: Como ocurre con todos los productos de proteínas administrados vía intravenosa, se pueden presentar reacciones de hipersensibilidad del tipo alérgico. Debe informarse a los pacientes de los signos iniciales de hipersensibilidad (que incluyen ronchas, urticaria generalizada, opresión en el pecho, jadeo e hipotensión) y anafilaxia. Si se presentan reacciones alérgicas o de anafilaxia, debe interrumpirse inmediatamente la administración de XYNTHA® y proporcionar manejo médico apropiado, que puede incluir el tratamiento para choque. Si alguno de los síntomas descritos ocurre, debe aconsejarse a los pacientes para que descontinúen el medicamento y contacten al médico y/o a un centro de urgencias dependiendo del tipo y la severidad de la reacción, está escrito al inicio de la fase. Precauciones: En los pacientes que reciben productos que contienen el factor VIII de la coagulación se pueden desarrollar anticuerpos neutralizantes de su actividad (inhibidores). Como sucede con todos los productos que contienen el factor VIII de la coagulación, se debe controlar a los pacientes para determinar si se presenta el desarrollo de inhibidores que deben ser titulados en Unidades Bethesda utilizando las pruebas biológicas apropiadas. Si no se obtienen los niveles de actividad plasmática esperados del factor VIII o si no se controla la hemorragia con una dosis apropiada, se deberá realizar examen para determinar si se encuentra presente un inhibidor del factor VIII. Estos inhibidores usualmente son inmunoglobulinas IgG que se dirigen contra la actividad procoagulante del factor VIII que se identifican en UB utilizando el ensayo Bethesda. El riesgo de desarrollar inhibidores se correlaciona con la exposición al factor VIII antihemofílico, este riesgo es mayor dentro de los primeros 20 días de exposición. Los inhibidores son comunes en pacientes no tratados previamente y se han observado en pacientes tratados previamente con productos del factor VIII. Se recomienda que, cuando sea posible, todas las veces que XYNTHA® se administre a los pacientes, se documente el nombre y número de lote del producto. Embarazo: No se han realizado estudios de reproducción animal con XYNTHA®. Debido a la ocurrencia poco frecuente de hemofilia A en mujeres, no se encuentra disponible experiencia relacionada con la utilización del factor VIII durante el embarazo. Por tanto, los productos del factor VIII se deben administrar a mujeres embarazadas únicamente si está claramente indicado. Lactancia: No se han realizado con XYNTHA® estudios de reproducción animal. Se desconoce si el medicamento se elimina en la leche materna. Debido a la ocurrencia poco frecuente de hemofilia A en mujeres, no se encuentra disponible experiencia relacionada con la utilización de productos del factor VIII durante la lactancia. Por lo tanto, los productos del factor VIII debe administrarse a mujeres que están lactando únicamente si está claramente indicado. Uso pediátrico: Se estudió la seguridad de XYNTHA® en niños y adolescentes previamente tratados (n=18, 12-16 años de edad en el estudio principal y n=49, 7-16 años de edad en el estudio de respaldo). En el estudio principal, los datos sobre eventos adversos de pacientes con ≤ 16 años de edad se compararon con los datos de pacientes de más 16 años de edad. Dieciocho (18) pacientes ≤ 16 años de edad y 76 de > 16 años de edad. El nivel de exposición en ambos grupos de pacientes fue similar. Los eventos adversos que surgieron durante el tratamiento fueron similares en severidad e incidencia en los dos grupos de edad. Ensayos clínicos que evalúan la utilización de moroctocog alfa (AF-CC) en PUP (estudio 4434), con niños menores de 6 años de edad (estudio 313) y niños menores de 12 años de edad (estudio 4433) se encuentran actualmente en curso. XYNTHA® puede utilizarse de la misma forma que se utilizaba el producto predecesor ReFacto, debido a que son bioquímicamente comparables y ha demostrado características farmacocinéticas similares con el producto predecesor ReFacto. Se ha estudiado la seguridad y eficacia del producto predecesor ReFacto en niños y adolescentes previamente tratados (n = 31, 5-18 años de edad) y en neonatos, infantes y niños no tratados previamente (n=101, < 1-52 meses de edad). Uso geriátrico: Los estudios clínicos de XYNTHA® no incluyeron sujetos de 65 o más años de edad. En general, se debe individualizar la selección de la dosis para los pacientes ancianos.
Interacciones.
No se conocen interacciones de los productos del factor VIII de la coagulación recombinante con otros medicamentos. Interacciones con pruebas de laboratorio y otras pruebas de diagnostico: No aplica.
Sobredosificación.
No se han reportado síntomas de sobredosis con productos del factor VIII de la coagulación.
Presentación.
XYNTHA Liofilizado con para solución inyectable 250 UI, con solvente. XYNTHA Liofilizado con para solución inyectable 500 UI, con solvente. XYNTHA Liofilizado con para solución inyectable 1000 UI, con solvente. XYNTHA Liofilizado con para solución inyectable 2000 UI, con solvente.


 Cambiar de país
Cambiar de país