MabThera®
ROCHE
Agente antineoplásico. Código ATC: L01XC02.
Composición.
Principio activo: rituximab. MabThera es un líquido incoloro, límpido, estéril, sin conservantes y apirógeno, presentado en viales monodosis. Viales monodosis. Viales con 100 mg/10 ml o 500 mg/50 ml. Excipientes: citrato sódico, polisorbato 80, cloruro sódico, hidróxido sódico, ácido clorhídrico y agua para inyectables. Forma farmacéutica: Concentrado para solución para infusión. Vía de administración: Infusión intravenosa (i.v). Declaración de esterilidad / radiactividad: Producto estéril.
Farmacología.
Propiedades farmacodinámicas: Mecanismo de acción: Rituximab es un anticuerpo monoclonal quimérico murino/humano, que se une específicamente al antígeno transmembranario CD20. Este antígeno se halla en los linfocitos pre-B y B maduros, pero no en las células madre hematopoyéticas, las células pro-B, las células plasmáticas fisiológicas ni en otros tejidos fisiológicos. El antígeno CD20 se expresa en más del 95% de todos los LNH de células B. Este antígeno no se internaliza tras la unión al anticuerpo ni se excreta de la superficie celular al medio circundante. El CD20 no circula en el plasma como antígeno libre, de modo que no compite por la unión a los anticuerpos. Rituximab se une al antígeno CD20 en los linfocitos B y desencadena reacciones inmunológicas mediadoras de la lisis de las células B. Mecanismos posibles de la lisis celular son la citotoxicidad dependiente del complemento (CDC), la citotoxicidad dependiente de anticuerpos (ADCC) y la inducción de apoptosis. Estudios in vitro han demostrado que el rituximab sensibiliza líneas de linfomas de células B humanas farmacorresistentes a los efectos citotóxicos de determinados quimioterápicos. El recuento de células B periféricas descendió hasta valores inferiores a la normalidad tras la primera dosis de MabThera. En los pacientes tratados de neoplasias hematológicas, la recuperación de la cifra de células B comenzó dentro de los 6 meses siguientes al inicio del tratamiento, normalizándose al cabo de 9-12 meses de concluido éste. En los pacientes con artritis reumatoide, la duración de la depleción de células B varió de unos a otros. La mayoría de ellos continuaron recibiendo tratamiento antes de la recuperación plena de las células B. En los pacientes con VAA, la cifra de células B CD19 en sangre periférica disminuyó a menos de 10 células/ml después de las dos primeras infusiones de rituximab y en la mayoría de los pacientes se mantuvieron en ese nivel a lo largo de los 6 meses. El resultado de la determinación de HAMA (anticuerpos humanos antimurinos) no fue positivo en ninguno de los 67 pacientes evaluados. De los 356 pacientes con LNH en los que se evaluaron los anticuerpos humanos antiquiméricos (HACA), el 1,1% (4 pacientes) arrojó un resultado positivo. Ensayos clínicos / Eficacia: Linfomas no hodgkinianos de bajo grado o foliculares: Monoterapia: Tratamiento inicial, 4 dosis a intervalos semanales: En el estudio clínico fundamental, 166 pacientes con LNH de bajo grado o folicular de células B recidivante o quimiorresistente recibieron 375 mg/m2 de MabThera en infusión i.v. semanal hasta completar cuatro dosis. La tasa global de respuesta (TGR) en la población con intención de tratar (ITT) fue del 48% (IC del 95%: 41-56%), con un 6% de respuestas completas (RC) y un 42% de respuestas parciales (RP). La mediana proyectada del tiempo hasta la progresión (TTP) en los pacientes respondedores fue de 13,0 meses. En un análisis de subgrupos, la TGR fue mayor en los pacientes con los subtipos histológicos B, C y D de la International Working Formulation (IWF) que en los que presentaban el subtipo A (58% y 12%, respectivamente), en los pacientes cuya lesión más grande era < 5 cm frente a aquellos con un diámetro máximo de las lesiones > 7 cm (53% y 38%, respectivamente) y, asimismo, mayor en los recidivantes con enfermedad quimiosensible que en los recidivantes con enfermedad quimiorresistente (definida como duración de la respuesta < 3 meses) (50% y 22%, respectivamente). La TGR fue del 78% en los pacientes que habían recibido previamente un autotrasplante de médula ósea (ATMO) frente al 43% en los pacientes sin ATMO. No se apreciaron efectos estadísticamente significativos (prueba exacta de Fisher) sobre la respuesta a MabThera para las siguientes variables: edad, sexo, grado del linfoma, diagnóstico inicial, presencia o ausencia de afección voluminosa, LDH normal o elevada, presencia de enfermedad extraganglionar. Se apreció una correlación estadísticamente significativa entre las tasas de respuesta y la afectación medular. Respondieron al tratamiento el 40% de los pacientes con afectación de la médula ósea frente a un 59% de los pacientes sin afectación de la misma (p = 0,0186). Este dato no se vio confirmado por un análisis de regresión logística escalonada, en el que se identificaron los siguientes factores pronóstico: tipo histológico, positividad bcl-2 en situación basal, resistencia a la última quimioterapia recibida y enfermedad voluminosa. Tratamiento inicial, 8 dosis a intervalos semanales: En un estudio clínico multicéntrico con un solo grupo, 37 pacientes con LNH de bajo grado o folicular de células B recidivante o quimiorresistente recibieron 375 mg/m2 de MabThera en infusión i.v. semanal hasta completar ocho dosis. La TGR fue del 57% (IC del 95%: 41-73%; RC: 14%; RP: 43%), con una mediana proyectada del tiempo hasta la progresión de 19,4 meses (intervalo: 5,3-38,9 meses) en los pacientes respondedores. Tratamiento inicial, enfermedad voluminosa, 4 dosis a intervalos semanales: En los datos combinados de tres estudios, 39 pacientes con LNH de bajo grado o folicular de células B recidivante o quimiorresistente, con afección voluminosa (lesión ≥ 10 cm de diámetro), recibieron 375 mg/m2 de MabThera en infusión i.v. semanal hasta completar cuatro dosis. La TGR fue del 36% (IC del 95%: 21-51%; RC: 3%; RP: 33%), con una mediana del TTP de 9,6 meses (intervalo: 4,5-26,8 meses) en los pacientes respondedores. Retratamiento, 4 dosis a intervalos semanales: En un estudio multicéntrico con un solo grupo, 58 pacientes con LNH de bajo grado o folicular de células B recidivante o quimiorresistente que habían tenido una respuesta clínica objetiva a una tanda anterior de MabThera recibieron retratamiento con 375 mg/m2 de MabThera en infusión i.v. semanal hasta completar cuatro dosis. Tres de ellos habían recibido dos tandas de MabThera antes del reclutamiento, por lo que recibieron una tercera tanda en el estudio. Dos pacientes recibieron dos veces retratamiento en el estudio. La TGR de los 60 retratamientos del estudio fue del 38% (IC del 95%: 26-51%; RC: 10%; RP: 28%), con una mediana proyectada del TTP de 17,8 meses (intervalo: 5,4- 26,6 meses) en los pacientes respondedores. Este resultado aventaja al TTP obtenido tras la tanda anterior de MabThera (12,4 meses). En asociación con quimioterapia: Tratamiento inicial: En un estudio abierto y aleatorizado, un total de 322 pacientes con linfoma folicular no tratado previamente recibieron, bien quimioterapia CVP (750 mg/m2 de ciclofosfamida, 1,4 mg/ m2 de vincristina hasta un máximo de 2 mg el día 1 y 40 mg/ m2/día de prednisolona los días 1 a 5) cada 3 semanas durante 8 ciclos, bien 375 mg/ m2 de MabThera con CVP (R-CVP). MabThera se administró en el primer día de cada ciclo de tratamiento. Un total de 321 pacientes (R-CVP: 162; CVP: 159) recibieron tratamiento y fueron evaluados para determinar la eficacia. La mediana de seguimiento de los pacientes fue de 53 meses. En la variable principal de valoración, el tiempo hasta el fracaso terapéutico, R-CVP proporcionó un beneficio significativo sobre CVP (27 frente a 6,6 meses, p < 0,0001, prueba de rangos logarítmicos). La proporción de pacientes con respuesta tumoral (RC, RC no confirmada, RP) fue significativamente más alta (p < 0,0001, prueba de c cuadrado) en el grupo de R- CVP (80,9%) que en el grupo de CVP (57,2%). El tratamiento con R-CVP prolongó significativamente el tiempo hasta la progresión de la enfermedad o la muerte en comparación con CVP (33,6 y 14,7 meses, respectivamente; p < 0,0001, prueba de rangos logarítmicos). La mediana de la duración de la respuesta fue de 37,7 meses en el grupo de R-CVP y de 13,5 meses en el grupo de CVP (p < 0,0001, prueba de rangos logarítmicos). La diferencia de supervivencia global entre los grupos de tratamiento revelaba un fuerte beneficio clínico (p = 0,029, prueba de rangos logarítmicos con estratificación por centros): la tasa de supervivencia a los 53 meses fue del 80,9% en el grupo de R-CVP y del 71,1% en el grupo de CVP. Los resultados de otros tres estudios aleatorizados con MabThera en asociación con una quimioterapia distinta a CVP (CHOP, MCP, CHVP/interferón alfa) también han demostrado mejoras significativas de la tasa de respuesta, los parámetros dependientes del tiempo y la supervivencia global. En la Tabla 4 se resumen los resultados principales de los cuatro estudios.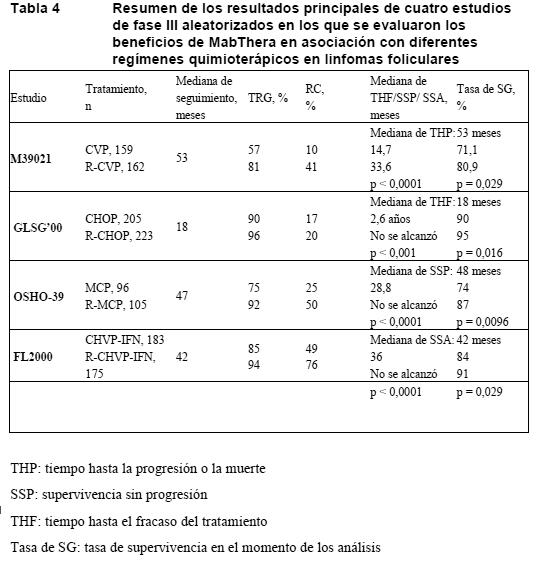
Terapia de mantenimiento: LNH foliculares no tratados previamente: En un estudio de fase III prospectivo, abierto, multicéntrico e internacional, 1.193 pacientes con linfoma folicular avanzado no tratado previamente recibieron terapia de inducción con CHOP (n = 881), R-CVP (n = 268) o R-FCM (n = 44) según la elección del investigador. Un total de 1.078 pacientes respondieron a la terapia de inducción; de ellos, 1.018 se aleatorizaron a un grupo de terapia de mantenimiento con MabThera (n = 505) o un grupo de observación (n = 513). Los dos grupos de tratamiento estaban bien equilibrados en cuanto a características basales y estado clínico. La terapia de mantenimiento con MabThera consistió en una infusión única de MabThera de 375 mg/ m2 de SC cada 2 meses hasta la progresión de la enfermedad o durante un máximo de dos años. Al cabo de una mediana de observación de 25 meses desde la aleatorización, la terapia de mantenimiento con MabThera se había traducido en una mejora clínicamente importante y estadísticamente significativa de la variable principal de valoración, la SSP evaluada por el investigador, en comparación con la ausencia de terapia de mantenimiento en pacientes con LNH folicular no tratado previamente. Un Comité institucional de revisión confirmó la mejora de la SSP (Tabla 5). Una ventaja significativa de la terapia de mantenimiento con MabThera se observó también en las variables secundarias de valoración: supervivencia sin acontecimientos (SSA), tiempo hasta el próximo tratamiento del linfoma (TPTL), tiempo hasta la próxima quimioterapia (TPQ) y tasa global de respuesta (TGR) (Tabla 5).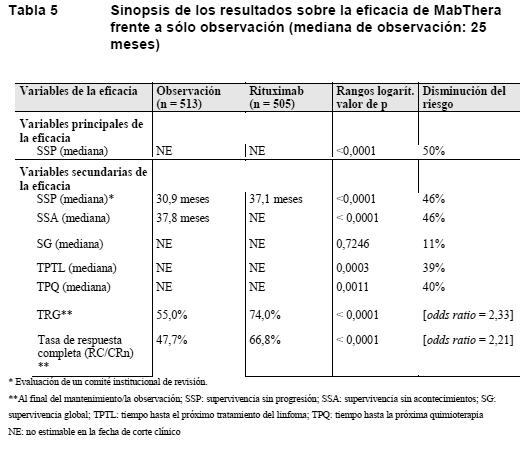
La terapia de mantenimiento con MabThera proporcionó un beneficio clínico sistemático en todos los subgrupos ensayados (sexo [masculino, femenino], edad [ < 60 años, > = 60 años], índice FLIPI [1, 2 o 3], terapia de inducción [R-CHOP, R-CVP o R-FCM]) e independientemente de la calidad de la respuesta a la terapia de inducción (RC o RP). LNH folicular recidivante/refractario: En un estudio de fase III, prospectivo, abierto, internacional y multicéntrico, 465 pacientes con LNH folicular recidivante/refractario fueron distribuidos aleatorizadamente en una primera fase a recibir terapia de inducción con CHOP (n = 231) o MabThera + CHOP (R-CHOP, n = 234). Los dos grupos de tratamiento estaban bien equilibrados en cuanto a características basales y estado clínico. En una segunda fase, se aleatorizó a un total de 334 pacientes con remisión completa o parcial tras la terapia de inducción para recibir terapia de mantenimiento con MabThera (n = 167) o permanecer en observación (n = 167). La terapia de mantenimiento con MabThera consistió en una infusión de MabThera de 375 mg/ m2 cada 3 meses hasta la progresión de la enfermedad o durante un máximo de dos años. En el análisis final de la eficacia se incluyó a todos los paciente aleatorizados a ambas partes del estudio. Después de una mediana de observación de 31 meses de los pacientes aleatorizados en la fase de inducción, los resultados de los pacientes con LNH recidivante o refractario del grupo de R-CHOP mejoraron significativamente en comparación con el grupo de quimioterapia CHOP (ver Tabla 6).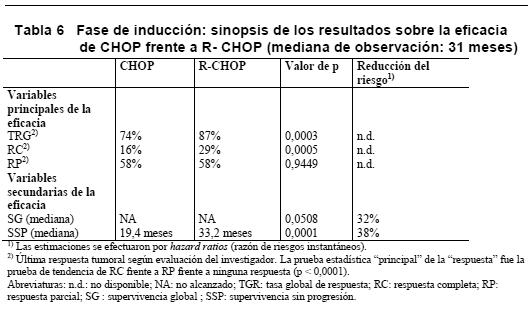
Para los pacientes aleatorizados a la fase de mantenimiento del estudio, la mediana de observación fue de 28 meses desde la aleatorización para el mantenimiento. La terapia de mantenimiento con MabThera se tradujo en una mejora clínicamente importante y estadísticamente significativa de la variable de valoración principal, SSP (tiempo entre la aleatorización para el mantenimiento y la recaída, la progresión de la enfermedad o la muerte), en comparación con el grupo de sólo observación (p < 0,0001, prueba de rangos logarítmicos). La mediana de la SSP fue de 42,2 meses en el grupo de mantenimiento con MabThera frente a 14,3 meses en el grupo de observación. Aplicando un análisis de regresión de Cox, el riesgo de enfermedad progresiva o fallecimiento disminuyó en un 61% en el grupo de terapia de mantenimiento con MabThera en comparación con el grupo de observación (IC del 95 %, 45-72%). La tasa estimada de supervivencia sin progresión a los 12 meses según el método de Kaplan-Meier era del 78% en el grupo de mantenimiento con MabThera frente al 57% en el de observación. Un análisis de la supervivencia global confirmó los beneficios significativos del mantenimiento con MabThera sobre la observación (p = 0,0039, prueba de rangos logarítmicos). La terapia de mantenimiento con MabThera redujo el riesgo de muerte en un 56% (IC del 95 %: 22- 75%). La mediana de la duración hasta un nuevo tratamiento del linfoma fue significativamente más larga en el grupo de terapia de mantenimiento con MabThera que en el de observación (38,8 frente a 20,1 meses; p < 0,0001, prueba de rangos logarítmicos). El riesgo de empezar un nuevo tratamiento disminuyó en un 50% (IC del 95%: 30-64%). En los pacientes con una RC/RCn (respuesta completa no confirmada) como mejor respuesta durante la terapia de inducción, la terapia de mantenimiento con MabThera prolongó significativamente la mediana de la supervivencia sin enfermedad (SSE) en comparación con el grupo de observación (53,7 frente a 16,5 meses; p = 0,0003, prueba de rangos logarítmicos) (Tabla 7). El riesgo de recaída de los pacientes con respuesta completa fue del 67% (IC del 95%: 39-82%).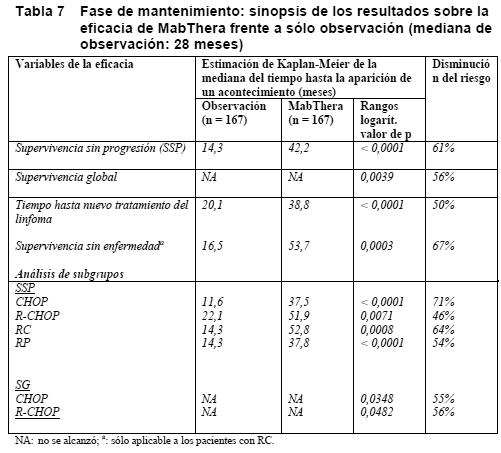
Los beneficios de la terapia de mantenimiento con MabThera se confirmaron en todos los subgrupos analizados, con independencia del régimen de inducción (CHOP o R-CHOP) o la calidad de la respuesta a la terapia de inducción (RC o RP) (Tabla 7). La terapia de mantenimiento con MabThera prolongó significativamente la mediana de la SSP tanto en los pacientes respondedores a la terapia de inducción con CHOP (mediana de SSP: 37,5 frente a 11,6 meses, p < 0,0001) como en los respondedores a la terapia de inducción con R-CHOP (mediana de SSP: 51,9 frente a 22,1 meses, p < 0,0071). La terapia de mantenimiento con MabThera también proporcionó un beneficio clínico significativo en la supervivencia global de los pacientes respondedores tanto a CHOP como a R-CHOP en la fase de inducción del estudio. La terapia de mantenimiento con MabThera se tradujo en beneficios sistemáticos para todos los subgrupos de estudio [sexo (masculino, femenino), edad (≤ 60 años, > 60 años), estadio (III, IV), estado general según la OMS (0 frente a > 0), síntomas B (ausentes, presentes), afectación de la médula ósea (no frente a sí), IPI (0-2 frente a 3-5), FLIPI (0-1, frente a 2 frente a 3-5), número de localizaciones extranodales (0-1 frente a > 1), número de localizaciones nodales ( < 5 frente a ≥ 5), número de regímenes anteriores (1 frente a 2), mejor respuesta a tratamiento anterior (RC/RP frente a sin cambios/EP), hemoglobina ( < 12 g/dl frente a ≥ 12 g/dl), b2-microglobulina ( < 3 mg/l frente a ≥ 3 mg/l), LDH (elevada, no elevada), salvo en el pequeño subgrupo de pacientes con afección voluminosa. Linfomas no hodgkinianos difusos de células B grandes: En un ensayo abierto y aleatorizado, un total de 399 pacientes ancianos no tratados previamente (edad: 60-80 años) que padecían linfomas difusos de células B grandes recibieron quimioterapia CHOP estándar (ciclofosfamida: 750 mg/m2; doxorrubicina: 50 mg/m2; vincristina 1,4 mg/m2 hasta un máximo de 2 mg el día 1, y prednisolona: 40 mg/ m2/día los días 1-5) cada 3 semanas, durante 8 ciclos, o bien MabThera 375 mg/ m2 + CHOP (R-CHOP). MabThera se administró en el primer día de cada ciclo de tratamiento. En el análisis final de la eficacia se incluyó a todos los pacientes aleatorizados (CHOP: 197; R-CHOP: 202), y la mediana de la duración del seguimiento fue de aproximadamente 31 meses. Los dos grupos de tratamiento estaban bien equilibrados en cuanto a características basales y estado clínico de los pacientes. En el análisis final se confirmó que en el grupo de R-CHOP había aumentado significativamente la duración de la supervivencia sin acontecimientos (la variable principal de la eficacia, siendo los acontecimientos la muerte, la recaída o la progresión del linfoma, o la instauración de un nuevo tratamiento del linfoma) (p = 0,0001). Las estimaciones de Kaplan-Meier de la mediana de supervivencia sin acontecimientos eran de 35 meses en el grupo de R-CHOP frente a 13 meses en el grupo de CHOP, lo que representa una reducción del riesgo del 41%. A los 24 meses, las estimaciones de la supervivencia global eran del 68,2% en el grupo de R-CHOP frente al 57,4% en el grupo de CHOP. Un análisis ulterior de la duración de la supervivencia global, realizado con una mediana del seguimiento de 60 meses, confirmó la ventaja de R-CHOP sobre CHOP (p = 0,0071), lo que representa una reducción del riesgo del 32%. El análisis de todos los parámetros secundarios (tasas de respuesta, supervivencia sin progresión, supervivencia sin enfermedad, duración de la respuesta) corroboró el efecto del tratamiento con R-CHOP comparado con el régimen CHOP. La tasa de respuesta completa después del ciclo 8 fue del 76,2% en el grupo de R-CHOP y del 62,4% en el grupo de CHOP (p = 0,0028). El riesgo de progresión de la enfermedad disminuyó en un 46%, y el riesgo de recaída, en un 51%. El índice de riesgo para la supervivencia sin acontecimientos y la supervivencia global (R-CHOP en relación con CHOP) era menor de 0,83 y 0,95, respectivamente, en todos los subgrupos de pacientes (sexo, edad, IPI ajustado por la edad, estadio de Ann Arbor, ECOG, beta2-microglobulina, LDH, albúmina, síntomas B, enfermedad voluminosa, enfermedad extranodal, afectación medular). Según el IPI ajustado por la edad, el régimen R-CHOP se acompañó de mejoras en los resultados tanto en los pacientes de alto riesgo como en los de bajo riesgo. Leucemia linfocítica crónica no tratada previamente y recidivante/refractaria: En dos ensayos abiertos y aleatorizados, un total de 817 pacientes con LLC no tratados previamente y 552 con LLC recidivante/refractaria recibieron quimioterapia FC (fludarabina: 25 mg/m2; ciclofosfamida: 250 mg/m2; días 1-3) cada 4 semanas, durante 6 ciclos, o MabThera + FC (R-FC). MabThera se administró en una dosis de 375 mg/ m2 durante el primer ciclo un día antes de la quimioterapia y de 500 mg/ m2 el día 1 de cada ciclo siguiente. Se analizó la eficacia en un total de 810 pacientes (R-FC: 403; FC: 407) del estudio del tratamiento de primera línea (Tabla 8) y 552 pacientes (R-FC: 276; FC: 276) del estudio de LLC recidivante/refractaria (Tabla 9). La supervivencia sin progresión, variable de valoración principal, fue de 40 meses (mediana) en el grupo de R-FC y de 32 meses en el grupo de FC (p < 0,0001, prueba de rangos logarítmicos). El análisis de la supervivencia global reveló una supervivencia mejorada en el grupo de R-FC (p = 0,0427, prueba de rangos logarítmicos); ahora bien, se requiere un seguimiento más largo para confirmar esta observación. Considerando el riesgo basal de enfermedad, en casi todos los subgrupos analizados se observaron sistemáticamente beneficios en la supervivencia sin progresión.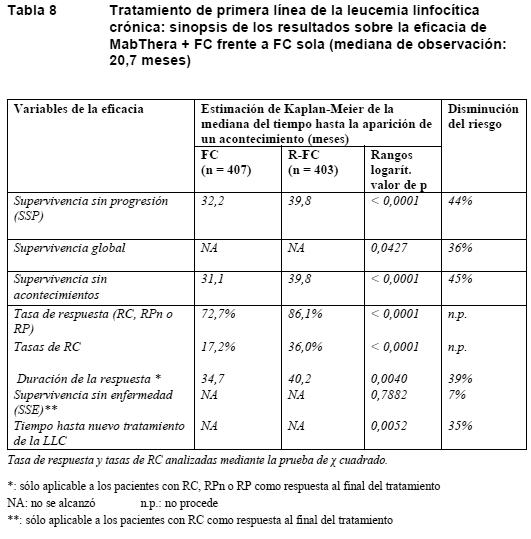
En el estudio de la LLC recidivante/refractaria, la mediana de la supervivencia sin progresión, variable principal de valoración, fue de 30,6 meses en el grupo de R-FC y de 20,6 meses en el de FC (p < 0,0002, prueba de rangos logarítmicos). Considerando el riesgo basal de enfermedad, en casi todos los subgrupos analizados se observaron beneficios en la supervivencia sin progresión. Se describió una mejora ligera, pero no significativa, de la supervivencia global en el grupo de R-FC comparado con el de FC.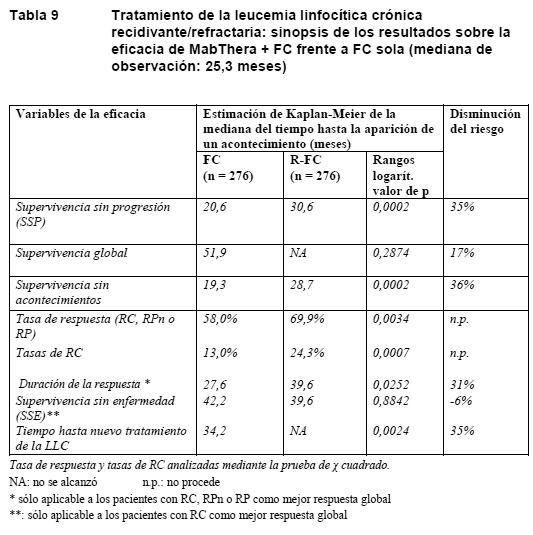
Los resultados de otros estudios de apoyo con MabThera en asociación con otros regímenes quimioterápicos (incluidos CHOP, FCM, PC y PCM, bendamustina y cladribina) para el tratamiento de la LLC también han demostrado altas tasas globales de respuesta y tasas prometedoras de SSP sin un aumento relevante de la toxicidad. Artritis reumatoide: La eficacia y la seguridad de MabThera para aliviar los signos y síntomas de la artritis reumatoide quedó demostrada en tres estudios multicéntricos, aleatorizados, de doble-ciego y controlados. El estudio 1 fue un ensayo clínico comparativo de doble-ciego, en el que participaron 517 pacientes que no habían respondido adecuadamente a uno o más inhibidores del TNF o no los toleraban. Los pacientes seleccionables presentaban artritis reumatoide activa grave, diagnosticada con arreglo a los criterios del Colegio Americano de Reumatología (ACR). La variable principal de valoración era el porcentaje de pacientes con una respuesta ACR20 en la semana 24. Los pacientes recibieron dos infusiones i.v. de 1.000 mg de MabThera, cada una de ellas tras una infusión i.v. de 100 mg de metilprednisolona y separadas por un intervalo de 15 días. Todos los pacientes recibieron concomitantemente metotrexato oral (10-25 mg/semana), así como 60 mg de prednisolona oral los días 2-7 y 30 mg los días 8-14 tras la primera infusión. El seguimiento de los pacientes se prolongó más allá de la semana 24 para evaluar las variables de valoración a largo plazo, incluida una evaluación radiográfica a las 56 semanas. Durante este período, los pacientes podían recibir nuevas tandas de rituximab según el protocolo de un estudio de extensión abierto. El estudio 2 fue un ensayo clínico aleatorizado, de doble-ciego, con doble placebo, controlado y multifactorial, en el que se compararon dos dosis diferentes de rituximab administrado con o sin uno de dos regímenes de corticosteroides en infusión, en asociación con metotrexato semanalmente, en pacientes con artritis reumatoide activa que no habían respondido al tratamiento con al menos otros 5 FAMEs (fármacos antirreumáticos modificadores de la enfermedad). El estudio 3 fue un ensayo clínico de doble-ciego, con doble placebo y controlado, en el que se evaluó rituximab en monoterapia y en asociación con ciclofosfamida o metotrexato en pacientes con artritis reumatoide activa que no habían respondido antes a uno o más FAMEs. El grupo de comparación en los tres estudios recibió metotrexato semanalmente (10-25 mg/semana). Parámetros de actividad de la enfermedad: En los tres estudios, la administración de 2 dosis de 1.000 mg de rituximab incrementó la proporción de pacientes con una mejora del índice ACR de un 20% como mínimo en comparación con los pacientes tratados con metotrexato solo (Tabla 7). El efecto terapéutico fue similar independientemente de variables como factor reumatoide, edad, sexo, superficie corporal, raza, número de tratamientos anteriores o estado clínico de los pacientes. También se observó una mejoría clínica estadísticamente significativa de todos los componentes de la respuesta ACR (número de articulaciones doloridas e inflamadas, evaluación global del paciente y del médico, índice HAQ [medida de la incapacidad funcional], evaluación del dolor y proteína C-reactiva [mg/dl]).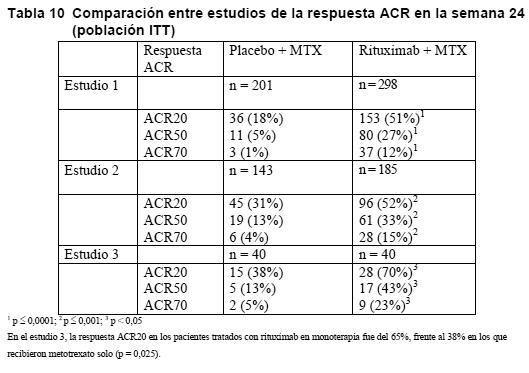
En los pacientes tratados con rituximab fue significativamente mayor el descenso del índice de actividad de la enfermedad (DAS28) que en los que recibieron metotrexato solo. Una respuesta EULAR buena o moderada la alcanzó un número significativamente mayor de pacientes tratados con rituximab que con metotrexato solo (Tabla 8).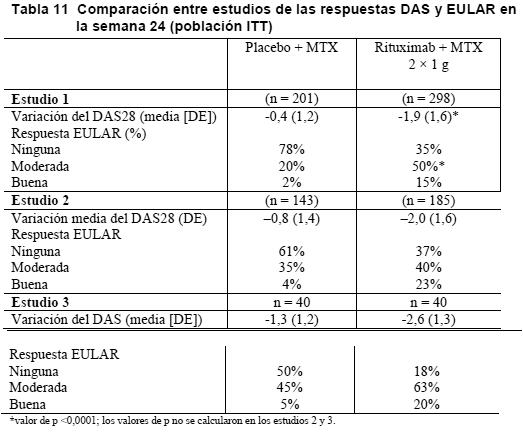
Respuesta radiográfica: En el estudio 1, realizado en pacientes que no habían respondido adecuadamente a uno o más a uno o más inhibidores del TNF o no los toleraban, el daño estructural articular se evaluó radiográficamente y se expresó como cambio del índice total de Sharp modificado y sus componentes, la puntuación de la erosión y la puntuación del estrechamiento del espacio articular. Al cabo de 56 semanas, en los pacientes del grupo rituximab + MTX era demostrable una progresión radiográfica significativamente menor que en los del grupo con metotrexato solo. La proporción de pacientes tratados con rituximab sin progresión de la erosión a las 56 semanas era también mayor (Tabla 9).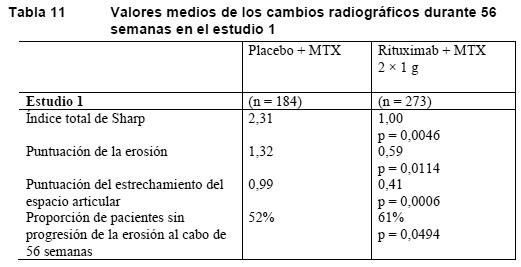
Parámetros de la calidad de vida: Los pacientes tratados con rituximab refirieron una mejora de todos los parámetros autoevaluados (cuestionarios HAQ-DI, FACIT-F y SF-36, Tablas 9 y 10). Se alcanzaron reducciones significativas en los índices de incapacidad (HAQ-DI) y fatiga (FACIT-F), así como una mejoría de los aspectos de salud física y mental del cuestionario SF-36 en los pacientes tratados con rituximab en comparación con los que recibieron metotrexato solo.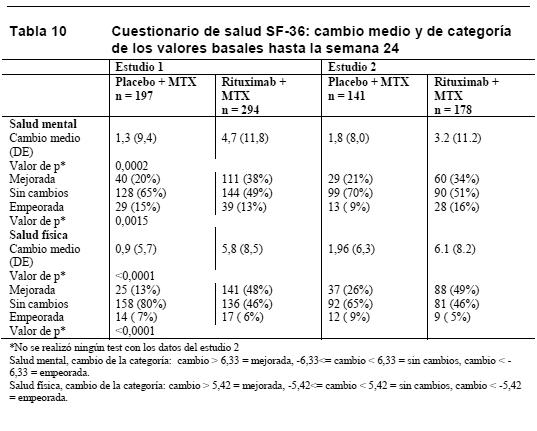
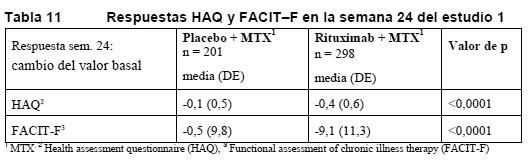
En la semana 24 y en los tres estudios, la proporción de pacientes tratados con rituximab con una mejoría clínicamente relevante en el HAQ-DI (definida como descenso de la puntuación total individual > 0,25) fue mayor que entre los pacientes tratados con metotrexato solo. Evaluaciones de laboratorio: En los estudios clínicos, aproximadamente el 10% de los pacientes con artritis reumatoide presentaban HACA. En la mayoría de los pacientes, la detección de HACA no se asoció a deterioro clínico o un mayor riesgo de reacciones a las infusiones siguientes. La presencia de HACA puede estar relacionada con un empeoramiento de las reacciones a la infusión o de reacciones alérgicas tras la segunda infusión de nuevas tandas; en raras ocasiones se ha observado fallo en la depleción de células B después de nuevas tandas de tratamiento. Entre los pacientes con un resultado positivo en el análisis del factor reumatoide (FR), en los tres estudios se produjeron descensos pronunciados de las concentraciones de FR tras el tratamiento con rituximab (intervalo: 45-64%, Figura 1).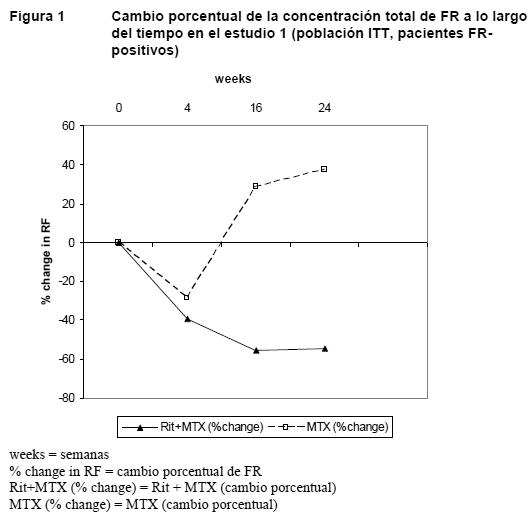
Las concentraciones plasmáticas de inmunoglobulinas totales, los recuentos totales de linfocitos y las cifras de leucocitos se mantuvieron en general dentro de límites normales tras la administración con MabThera, excepción hecha de una caída temporal del número de leucocitos en las primeras cuatro semanas tras el tratamiento. Los títulos de anticuerpos específicos IgG contra antígenos de paperas, rubéola, varicela, toxoide tetánico, gripe y Streptococcus pneumococci permanecieron estables durante 24 horas tras la exposición a MabThera en los pacientes con artritis reumatoide. Los efectos del rituximab en diversos biomarcadores se evaluaron en los pacientes participantes en un estudio clínico. En este subestudio se evaluó el impacto de una tanda única de rituximab en los niveles de marcadores bioquímicos, a saber: marcadores de inflamación (interleucina 6, proteína C-reactiva, proteína SAA [suero amiloide A], proteína S100 isotipos A8 y A9), autoanticuerpos (FR y anticuerpos antipéptidos cíclicos citrulinados) producción y recambio óseo (osteocalcina y péptido aminoterminal del procolágeno [P1NP]). El tratamiento con rituximab, en monoterapia o en asociación con metotrexato o ciclofosfamida, redujo los niveles de marcadores inflamatorios significativamente en comparación con el metotrexato solo durante las primeras 24 horas de seguimiento. Los niveles de marcadores de recambio óseo, osteocalcina y P1NP aumentaron significativamente con rituximab en comparación con metotrexato solo. Múltiples tandas de tratamiento: Terminado el período de estudio comparativo a doble-ciego de 24 semanas, los pacientes podían participar en un estudio abierto de seguimiento a largo plazo. Los pacientes recibieron nuevas tandas de MabThera según las necesidades de acuerdo con la evaluación por el médico de la actividad de la enfermedad y sin tener en cuenta la cifra de linfocitos B periféricos. El lapso de tiempo entre dos tandas variaba, pero la mayoría de los pacientes fueron tratados de nuevo a los 6-12 meses de finalizada la tanda anterior. La necesidad de retratamiento fue incluso menos frecuente en algunos pacientes. Cuando se continuó el tratamiento, la respuesta fue al menos de igual magnitud que tras la tanda inicial, como mostraba la variación del DAS28 (Figura 2).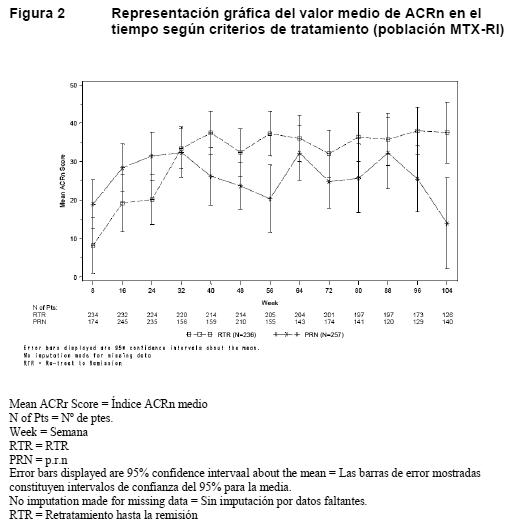
Vasculitis asociada a ANCA (VAA): Se reclutó y trató a un total de 197 pacientes con vasculitis asociada a anticuerpos anticitoplasma de neutrófilo (ANCA) gravemente activa en un estudio de no inferioridad controlado con tratamiento activo, aleatorizado, de doble-ciego y multicéntrico. Los pacientes tenía 15 o más años de edad y se les había diagnosticado granulomatosis de Wegener activa (75% de los pacientes) o poliangitis microscópica (PAM) (24%) según los criterios de la Conferencia de Consenso de Chapel Hill (el 1% de los pacientes presentaban una VAA de tipo desconocido). Se aleatorizó a los pacientes en la proporción 1:1 para recibir, bien ciclofosfamida oral diariamente (2 mg/kg/día) durante 3-6 meses, seguida de azatioprina, bien MabThera (375 mg/m2) una vez por semana durante 4 semanas. Los pacientes de ambos grupos recibieron 1.000 mg/día de metilprednisolona i.v. (u otro glucocorticoide en una dosis equivalente) durante 1-3 días, seguido de prednisona oral (1 mg/kg/día, sin sobrepasar los 80 mg/día). La retirada escalonada de la prednisona debía estar completada a los 6 meses de iniciado el tratamiento en estudio. La variable principal de valoración de los resultados era la remisión completa al cabo de 6 meses, definida como una puntuación de 0 en el índice BVAS/WG (Birmingham Vasculitis Activity Score for Wegener's Granulomatosis), y haberse retirado los glucocorticoides. El margen preestablecido de no inferioridad para la diferencia en los tratamientos era del 20%. El estudio demostró que MabThera no era inferior a la ciclofosfamida para una remisión completa a los 6 meses (Tabla 5). Además, de acuerdo con datos de control históricos, la tasa de remisión completa en el grupo de MabThera fue significativamente más alta que la tasa estimada de remisión completa en los pacientes con VAA grave no tratados o tratados sólo con glucocorticoides. La eficacia se observó tanto en los pacientes con VAA recién diagnosticada como en los que habían sufrido una recaída.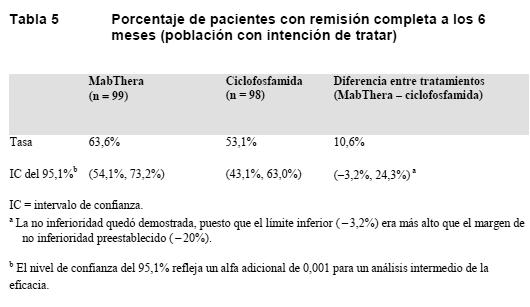
Propiedades farmacocinéticas: Distribución: Linfomas no hodgkinianos: De acuerdo con un análisis de farmacocinética poblacional en 298 pacientes con LNH que habían recibido una única o múltiples infusiones de rituximab en monoterapia o en asociación con CHOP, las estimaciones poblacionales típicas de aclaramiento no específico (CL1), aclaramiento específico (CL2) probablemente influido por las células B o la masa tumoral y volumen de distribución del compartimiento central (V1) eran de 0,14 l/día, 0,59 l/día y 2,7 l, respectivamente. La mediana estimada de la semivida de eliminación terminal del rituximab era de 22 días (intervalo: de 6,1 a 52 días). El recuento basal de células CD19-positivas y el tamaño de las lesiones tumorales contribuyeron en cierto grado a la variabilidad de los datos de CL2 del rituximab en los 161 pacientes que recibieron en infusión i.v. 4 dosis de 375 mg/ m2 a intervalos semanales. Los pacientes con cifras más altas de células CD19-positivas o con lesiones tumorales presentaban un CL2 más alto. Ahora bien, una gran parte de la variabilidad interindividual de CL2 se mantuvo tras la corrección realizada por el recuento de células CD19-positivas y el tamaño de lesión tumoral. V1 variaba en función de la superficie corporal y la quimioterapia CHOP. Esta variabilidad de V1 (27,1% y 19,0%), a la que contribuía la superficie corporal (de 1,53 a 2,32 m2) y el régimen CHOP concomitante, era relativamente pequeña. Edad, sexo, raza y estado general según la OMS no ejercían ningún efecto en la farmacocinética del rituximab. Este análisis sugiere que de un ajuste posológico de rituximab con cualquiera de las covariables no es de esperar una reducción significativa de su variabilidad farmacocinética. A 203 pacientes con LNH no tratados previamente con rituximab se les administraron 4 dosis de rituximab de 375 mg/m2 en infusión i.v. a intervalos semanales. La Cmáx media tras la cuarta infusión era de 486 mg/ml (intervalo: de 77,5 a 996,6 mg/ml). Las concentraciones séricas máxima y mínima de rituximab estaban en relación inversa con los valores basales de células B CD19-positivas circulantes y masa tumoral. La mediana de las concentraciones séricas en estado de equilibrio era mayor en los pacientes que respondían que en los no respondedores. Las concentraciones séricas eran más altas en los subtipos histológicos B, C y D de la IWF (International Working Formulation) que en los pacientes con el subtipo A. El rituximab era detectable en el suero de los pacientes 3- 6 meses después de concluido el último tratamiento. A 37 pacientes con LNH se les administraron 8 dosis de rituximab de 375 mg/m2 en infusión i.v. a intervalos semanales. La Cmáx aumentaba con cada nueva infusión, desde una media de 243 mg/ml (intervalo: 16-582 mg/ml) tras la primera infusión hasta 550 mg/ml (intervalo: 171-1.177 mg/ml) después de la octava. El perfil farmacocinético del rituximab administrado en 6 infusiones de 375 mg/m2 en asociación con 6 ciclos de quimioterapia CHOP era similar al registrado tras la administración de rituximab solo. Leucemia linfocítica crónica: Rituximab se administró a pacientes con LLC en infusión i.v., en una dosis de 375 mg/m2 en el primer ciclo, elevada en las 5 dosis siguientes a 500 mg/m2 cada ciclo, en asociación con fludarabina y ciclofosfamida. La Cmáx media (n = 15) fue de 408 mg/ml (intervalo de 97 a 764 mg/ml) tras la quinta infusión de 500 mg/m2. Artritis reumatoide: Tras la administración de dos infusiones i.v. de rituximab de 1.000 g cada una, separadas por dos semanas, la semivida media terminal fue de 20,8 días (intervalo: 8,58-35,9 días); el aclaramiento sistémico medio, de 0,23 l/día (intervalo: 0,091-0,67 l/día), y el volumen medio de distribución en equilibrio, de 4,6 litros (intervalo: 1,7-7,51 litros). Un análisis de farmacocinética poblacional de los mismos datos arrojó unos valores medios similares para el aclaramiento sistémico y la semivida: 0,26 l/día y 20,4 días, respectivamente. Un análisis farmacocinético poblacional reveló que la superficie corporal y el sexo eran las covariables más significativas para explicar la variabilidad interindividual de los parámetros farmacocinéticos. Tras el ajuste por la superficie corporal, el volumen de distribución era mayor y el aclaramiento más rápido en los sujetos masculinos que en los femeninos. Las diferencias farmacocinéticas asociadas al sexo no se consideraron clínicamente importantes y, por tanto, no es necesario ajustar la dosis. La farmacocinética del rituximab se evaluó en cuatro estudios tras dos dosis i.v. de 500 mg y 1.000 mg los días 1 y 15. En todos estos estudios, la farmacocinética del rituximab fue proporcional a la dosis en el intervalo limitado de dosis evaluadas. La Cmáx sérica media de rituximab tras la primera infusión se situó entre 157 y 171 mg/ml con 2 veces 500 mg y entre 298 y 341 mg/ml con 2 veces 1.000 mg. Tras la segunda infusión, la Cmáx media se situó entre 183 y 198 mg/ml con 2 veces 500 mg y entre 355 y 404 mg/ml con 2 veces 1.000 mg. La media de la semivida de eliminación terminal osciló entre 15 y 16,5 días con la dosis de 2 veces 500 mg y entre 17 y 21 días con la de 2 veces 1.000 mg. La Cmáx media fue un 16-19% más alta tras la segunda infusión con ambas dosis. La farmacocinética del rituximab se evaluó tras dos dosis i.v. de 500 mg y 1.000 mg los días 1 y 15 después del retratamiento en la segunda tanda. La Cmáx sérica media de rituximab tras la primera infusión se situó entre 170 y 175 mg/ml con 2 veces 500 mg y entre 317 y 370 mg/ml con 2 veces 1.000 mg. Tras la segunda infusión, la Cmáx fue de 207mg/ml con 2 veces 500 mg y de 377-386 mg/ml con 2 veces 1.000 mg. Tras la segunda infusión, la media de la semivida de eliminación terminal después de la segunda tanda fue de 19 días con la dosis de 2 veces 500 mg y de 21-22 días con la dosis de 2 veces 1.000 mg. Los parámetros farmacocinéticos del rituximab eran comparables con las dos tandas de tratamiento. Después de recibir la misma pauta de administración (2 dosis de 1.000 mg i.v., dos semanas aparte), los parámetros farmacocinéticos en los pacientes con una respuesta inadecuada al tratamiento con anti-TNF fue similar, con una concentración sérica media máxima de 369 mg/ml y una semivida terminal de 19,2 días. Vasculitis asociada a ANCA: De acuerdo con los resultados de un análisis poblacional de farmacocinética de 97 pacientes con VAA tratados con 375 mg/m2 de rituximab una vez por semana durante 4 semanas, la mediana estimada de la semivida de eliminación terminal fue de 23 días (extremos: 9 y 49 días). Los valores medios de aclaramiento y volumen de distribución del rituximab fueron de 0,313 l/día (extremos: 0,116 y 0,726 l/día) y 4,50 litros (extremos: 2,25 y 7,39 litros), respectivamente. Los parámetros farmacocinéticos del rituximab en los pacientes con VAA parecen similares a los registrados en los pacientes con AR (ver apartados anteriores). Eliminación: Ver Distribución. Farmacocinética en poblaciones especiales: No existen datos farmacocinéticos de pacientes con menoscabo hepático o renal.
Indicaciones.
Linfomas no hodgkinianos: MabThera está indicado para: el tratamiento de pacientes con linfoma no hodgkiniano (LNH) de bajo grado o folicular de células B CD20-positivas, recidivante o quimiorresistente;
Dosificación.
Instrucciones generales: MabThera debe administrarse en infusión intravenosa (i.v.), por una vía específica, en un entorno hospitalario con un equipo completo de reanimación inmediatamente disponible y bajo la estrecha vigilancia de un médico experimentado. La solución para infusión preparada no debe administrarse en infusión rápida o en bolo i.v. Como premedicación deben administrarse siempre un analgésico/antipirético (por ejemplo: paracetamol) y un antihistamínico (por ejemplo: difenhidramina) antes de cada infusión de MabThera. También debe considerarse la premedicación con glucocorticoides, particularmente si MabThera no se administra con quimioterapia que contenga esteroides (ver Dosis habitual). Dosis habitual: Linfomas no hodgkinianos de bajo grado o foliculares: Tratamiento inicial: La dosis recomendada de MabThera en monoterapia para pacientes adultos es de 375 mg/m2 de superficie corporal (SC), administrada en infusión i.v (ver Primera infusión e Infusiones siguientes), una vez por semana, durante 4 semanas. La dosis recomendada de MabThera en asociación con cualquier quimioterapia es de 375 mg/m2 de SC por ciclo, durante un total de: 8 ciclos con R-CVP (21 días/ciclo). 8 ciclos con R-MCP (28 días/ciclo). 8 ciclos con R-CHOP (21 días/ciclo); 6 ciclos si se alcanza la remisión completa después de 4 ciclos. 6 ciclos con R-CHVP-interferón (21 días/ciclo). MabThera debe administrarse el día 1 de cada ciclo de quimioterapia, tras la administración i.v. del componente glucocorticoide de la quimioterapia (si procede). Retratamiento tras una recaída: Se ha vuelto a tratar con MabThera (375 mg/m2 de SC en infusión i.v. semanal, durante 4 semanas) a pacientes que habían respondido inicialmente a este medicamento (ver Retratamiento, 4 dosis a intervalos semanales). Terapia de mantenimiento: Los pacientes no tratados previamente que hayan respondido a la terapia de inducción pueden recibir terapia de mantenimiento con MabThera en una dosis de 375 mg/m2 de SC, una vez cada 2 meses, hasta la progresión de la enfermedad o durante un máximo de 2 años (12 infusiones). Los pacientes en recidiva o refractarios que hayan respondido a la terapia de inducción pueden recibir terapia de mantenimiento con MabThera en una dosis de 375 mg/m2 de SC, una vez cada 3 meses, hasta la progresión de la enfermedad o durante un máximo de 2 años. Linfomas no hodgkinianos difusos de células B grandes: MabThera debe utilizarse en asociación con el régimen CHOP (ciclofosfamida, doxorrubicina, prednisona y vincristina). La dosis recomendada de MabThera es de 375 mg/m2 de SC, administrada el día 1 de cada ciclo de quimioterapia de 8 ciclos tras las administración i.v. del componente glucocorticoide de CHOP. Los restantes componentes de la quimioterapia CHOP deben administrarse después de MabThera (ver Primera infusión e Infusiones siguientes). Leucemia linfocítica crónica: Para reducir el riesgo de síndrome de lisis tumoral, en los pacientes con LLC se recomienda la profilaxis con suficiente hidratación y la administración de uricostáticos desde 48 horas antes del comienzo del tratamiento. En los pacientes con LLC con un recuento linfocitario > 25 x 109/l se recomienda administrar i.v. 100 mg de prednisona/prednisolona poco antes de la infusión de MabThera para reducir la tasa y la gravedad de reacciones agudas a la infusión y/o el síndrome de liberación de citocinas. La dosis recomendada de MabThera en asociación con quimioterapia para pacientes no tratados previamente o en recidiva/refractarios es de 375 mg/m2 de SC, administrada el día 1 del primer ciclo, seguida de 500 mg/m2 de SC, administrada el día 1 de cada ciclo siguiente, hasta un total de 6 ciclos (ver Ensayos clínicos / Eficacia). La quimioterapia debe administrarse después de la infusión de MabThera. Primera infusión: Se recomienda una velocidad inicial de infusión de 50 mg/h, aumentable después a razón de 50 mg/h cada 30 minutos, hasta un máximo de 400 mg/h. Infusiones siguientes: Las infusiones siguientes de MabThera pueden comenzarse a una velocidad de 100 mg/h, aumentándose ésta a continuación a razón de 100 mg/h cada 30 minutos, hasta un máximo de 400 mg/h. Ajustes posológicos durante el tratamiento: No se recomienda reducir la dosis de MabThera. Cuando se administre MabThera en asociación con quimioterapia, deben aplicarse las reducciones posológicas habituales para los quimioterápicos. Artritis reumatoide (AR): Una tanda de MabThera consiste en dos infusiones i.v. de 1.000 mg. La dosis recomendada de MabThera es de 1.000 mg en infusión i.v., seguida dos semanas después de una segunda infusión i.v. de 1.000 mg. A las 24 semanas de una tanda debe evaluarse la necesidad de una nueva tanda, basándose el retratamiento en el retorno de una actividad residual o de la enfermedad a un nivel superior a 2,6 de la respuesta DAS28-VSG (tratamiento hasta remisión) (ver Ensayos clínicos / Eficacia en la AR). Antes de transcurridas 16 semanas no debe administrarse una nueva tanda. Los pacientes deben recibir 100 mg de metilprednisolona i.v. 30 minutos antes de cada infusión de MabThera para reducir la incidencia y la gravedad de las reacciones relacionadas con la infusión (ver Advertencias y precauciones). Primera infusión de cada tanda: Se recomienda una velocidad inicial de infusión de 50 mg/h, aumentable al cabo de 30 minutos a razón de 50 mg/h cada 30 minutos, hasta un máximo de 400 mg/h. Segunda infusión de cada tanda: Las infusiones siguientes de MabThera pueden comenzarse a una velocidad de 100 mg/h, aumentable a razón de 100 mg/h cada 30 minutos, hasta un máximo de 400 mg/h. Vasculitis asociada a ANCA (VAA): La dosis recomendada de MabThera para el tratamiento de la VAA es de 375 mg/m2 de superficie corporal, en infusión i.v., una vez por semana durante 4 semanas. Para tratar los síntomas graves de vasculitis se recomienda administrar metilprednisolona, en una dosis de 1.000 mg/día i.v. durante 1-3 días, en combinación con MabThera, seguido de prednisona oral, en una dosis de 1 mg/kg/día (no deben sobrepasarse los 80 mg/día, reducidos progresivamente lo antes posible según el estado clínico), durante el tratamiento con MabThera y después de éste. Primera infusión: Se recomienda una velocidad inicial de infusión de MabThera de 50 mg/h, aumentable después a razón de 50 mg/h cada 30 minutos, hasta un máximo de 400 mg/h. Infusiones siguientes: Las infusiones siguientes de MabThera pueden comenzarse a una velocidad de 100 mg/h, aumentándose ésta a continuación a razón de 100 mg/h cada 30 minutos, hasta un máximo de 400 mg/h. Se recomienda la prevención de la neumonía por Pneumocystis jiroveci en los pacientes con VAA durante el tratamiento con MabThera y después de éste. Pautas posológicas especiales: Niños y adolescentes: No se han estudiado la seguridad y la eficacia de MabThera en niños y adolescentes. Ancianos: No se requieren ajustes posológicos en los ancianos (edad: > 65 años).
Contraindicaciones.
MabThera está contraindicado en pacientes con antecedentes de alergia al rituximab, a cualquier otro componente del producto o a proteínas murinas.
Reacciones adversas.
Ensayos clínicos: Experiencia obtenida en estudios clínicos de hematooncología: En las tablas siguientes se resume la frecuencia de reacciones adversas (RA) notificadas en los estudios clínicos con MabThera en monoterapia o en asociación con quimioterapia. Estas RA se produjeron en estudios con un solo grupo o con una diferencia mínima del 2% frente al grupo de control en al menos uno de los principales estudios clínicos aleatorizados. Las RA se han categorizado en las tablas de acuerdo con la incidencia más alta registrada en cualquiera de los estudios clínicos principales. Las RA están enumeradas dentro de cada grupo de frecuencia por orden descendente de gravedad. Las frecuencias se definen como muy frecuente ≥ 1/10, frecuente ≥ 1/100 a < 1/10 o poco frecuente ≥ 1/1.000 a < 1/100. MabThera en monoterapia / terapia de mantenimiento: Las RA de la Tabla 1 se basan en los datos de estudios con un solo grupo en 356 pacientes con linfoma de bajo grado o folicular que recibieron semanalmente MabThera en monoterapia como tratamiento o retratamiento de un linfoma no hodgkiniano hasta 4 semanas en la mayoría de los casos, así como en los datos de 25 pacientes tratados con dosis distintas a 375 mg/m2 en número de cuatro dosis y hasta 500 mg/m2 en una dosis única en el marco de la investigación clínica de fase I (ver Ensayos clínicos/Eficacia). La tabla también contiene las RA basadas en los datos de 671 pacientes con linfoma folicular que recibieron MabThera como terapia de mantenimiento por espacio de hasta 2 años tras la respuesta a la terapia de inducción con CHOP o R- CHOP (ver Ensayos clínicos/Eficacia). Las RA se notificaron hasta 12 meses después de la monoterapia y hasta 1 mes después de la terapia de mantenimiento con MabThera.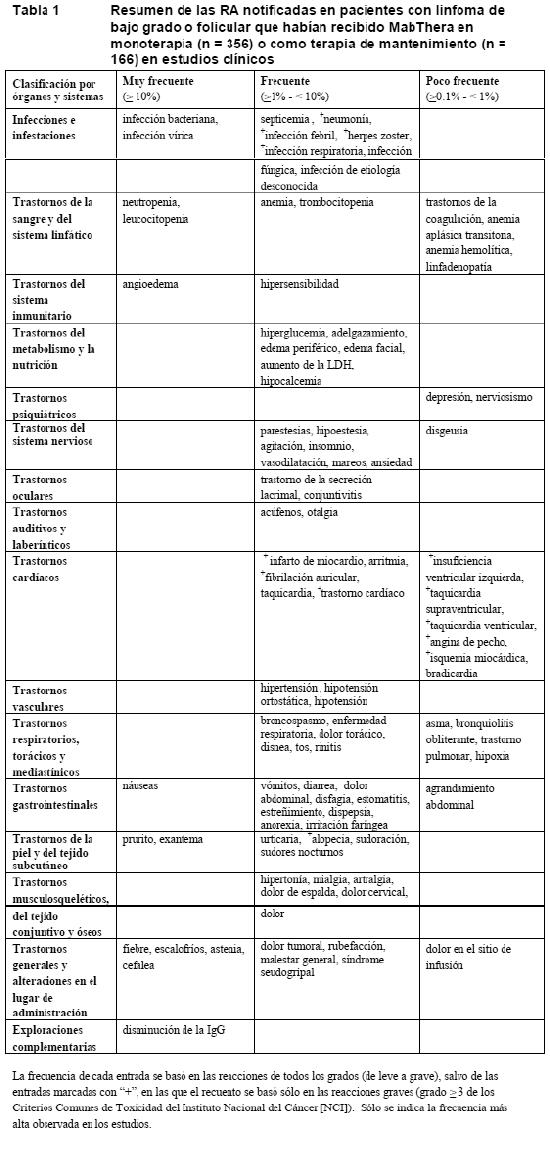
MabThera en asociación con quimioterapia en los LNH y la LLC: Las RA enumeradas en la Tabla 2 se basan en los datos del grupo de MabThera en estudios clínicos controlados que se produjeron además de los observados en la monoterapia y la terapia de mantenimiento o con una frecuencia mayor: 202 pacientes con linfoma difuso de células B grandes (LDCBG) tratados con R-CHOP, así como 234 y 162 pacientes con linfoma folicular tratados con R-CHOP o R-CVP, respectivamente, y 397 pacientes con LLC no tratados previamente y 274 con LLC recidivante/refractaria tratados con MabThera en asociación con fludarabina y ciclofosfamida (R-FC) (ver Ensayos clínicos/Eficacia).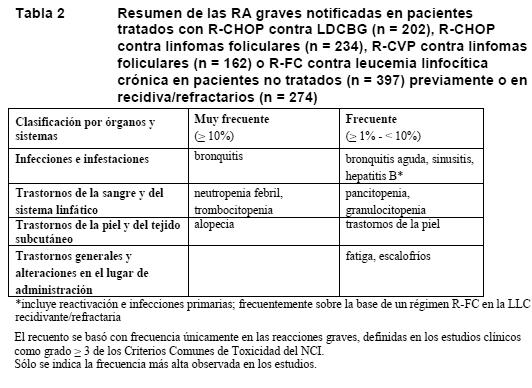
Los términos siguientes se han notificado como acontecimientos adversos; ahora bien, se notificaron con una incidencia similar ( < 2% de diferencia entre los grupos) o menor en los grupos de MabThera que en los de control: hematotoxicidad, infección neutropénica, infección urinaria, choque séptico, superinfección pulmonar, infección de un implante, septicemia estafilocócica, infección pulmonar, rinorrea, edema pulmonar, insuficiencia cardíaca, trastorno sensorial, trombosis venosa, mucositis (sin especificar), síndrome seudogripal, edema de las extremidades inferiores, fracción de eyección anormal, pirexia, deterioro del estado físico general, caída, fallo multiorgánico, trombosis venosa profunda, hemocultivo positivo, control inadecuado de diabetes mellitus. El perfil de seguridad de MabThera en asociación con otras quimioterapias (por ejemplo: CHOP, MCP, CHVP-IFN) es comparable al descrito para la asociación de MabThera y CVP, CHOP o FC en poblaciones equivalentes. Más información sobre reacciones adversas graves seleccionadas: Reacciones relacionadas con la infusión: Monoterapia durante 4 semanas: En asociación con la infusión de MabThera se han producido hipotensión, fiebre, escalofríos, urticaria, broncospasmo, angioedema, náuseas, fatiga, cefalea, prurito, disnea, rinitis, vómitos, rubefacción y dolor en el sitio de la enfermedad como parte de un complejo sintomático relacionado con la infusión. La mayoría de los pacientes experimentaron tales síntomas relacionados con la infusión con la primera infusión de MabThera (ver Advertencias). La incidencia de síntomas relacionados con la infusión descendió del 77% (7% de grados 3 o 4) con la primera infusión a aproximadamente un 30% (2% de grados 3 o 4) con la cuarta infusión y al 14% (ninguno de grados 3 o 4) con la octava. Terapia de mantenimiento (LNH) hasta 2 años: En el 41% de los pacientes se notificaron signos y síntomas no graves evocadores de una reacción relacionada con la infusión como trastornos generales (principalmente astenia, pirexia, enfermedad de tipo gripal y dolor) y en el 7% como trastornos del sistema inmunitario (hipersensibilidad). Reacciones graves relacionadas con la infusión se produjeron en < 1% de los pacientes. Politerapia (R-CVP en los LNH; R-CHOP en los LDCBG, R-FC en la LLC): Reacciones graves relacionadas con la infusión se produjeron en hasta un 12 % de todos los pacientes en el primer ciclo de rituximab en asociación con quimioterapia. La incidencia de reacciones graves relacionadas con la infusión había descendido a menos del 1% en el octavo ciclo. Los signos y síntomas coincidían con los observados durante la monoterapia (ver Advertencias y Reacciones adversas, MabThera en monoterapia), pero también comprendían dispepsia, exantema, hipertensión, taquicardia y rasgos del síndrome de lisis tumoral. Otras reacciones adversas notificadas con R-quimioterapia en casos aislados fueron infarto de miocardio, fibrilación auricular, edema pulmonar y trombocitopenia aguda reversible. Infecciones: Monoterapia durante 4 semanas: MabThera provocó la depleción de células B en un 70-80% de los pacientes, pero sólo en una minoría de los ellos se acompañó de una reducción de las inmunoglobulinas séricas. Se describieron episodios infecciosos, independientemente de la valoración causal, en el 30,3% de 356 pacientes: el 18,8% de ellos presentaban infecciones bacterianas; el 10,4%, infecciones víricas; el 1,4%, micosis, y el 5,9% restante, infecciones de etiología desconocida. En un 3,9% de los pacientes se describieron infecciones graves (grados 3 o 4), sepsis inclusive: el 1,4% durante el período de tratamiento, y el 2,5% restante, durante el período de seguimiento. Terapia de mantenimiento (LNH) hasta 2 años: La proporción de pacientes con infecciones de grados 1 a 4 fue del 25% en el grupo de observación y del 45% en el grupo de MabThera, siendo la infección grave (grados 3 o 4) en el 3% de los pacientes del grupo de observación y en el 11% de los que recibieron terapia de mantenimiento con MabThera. Las infecciones graves notificadas en ≥ 1% de los pacientes del grupo de MabThera fueron neumonía (2%), infección respiratoria (2%), infección febril (1%) y herpes zóster (1%). En una gran proporción de las infecciones (de todos los grados), el agente infeccioso no se especificó o aisló; ahora bien, cuando se especificó, los agentes infecciosos notificados con más frecuencia fueron bacterias (grupo de observación: 2%; grupo de MabThera: 10%), virus (grupo de observación: 7%; grupo de MabThera: 11%) y hongos (grupo de observación: 2%; grupo de MabThera: 4%). No se observó toxicidad acumulativa por lo que respecta a las infecciones notificadas en el período de mantenimiento de 2 años. Los datos de un estudio clínico de fase III incluían dos casos de LEMP en pacientes con LNH ocurridos tras la progresión de la enfermedad y el retratamiento (ver Advertencias). Politerapia (R-CVP en los LNH; R-CHOP en los LDCBG, R-FC en la LLC): En el estudio con R-CVP, la proporción total de pacientes con infecciones o infestaciones durante el tratamiento y los primeros 28 días siguientes a su terminación era comparable en los grupos de tratamiento (R-CVP: 33%; CVP: 32%). Las infecciones más frecuentes eran de las vías respiratorias superiores y se notificaron en el 12,3% de los pacientes tratados con R-CVP y el 16,4% de los que recibieron CVP; en la mayoría de los casos, las infecciones consistieron en nasofaringitis. Infecciones graves se notificaron en el 4,3% de los pacientes tratados con R-CVP y el 4,4% de los que recibieron CVP. En este estudio no se notificó ninguna infección que comportara peligro de muerte. En el estudio con R-CHOP, la incidencia global de infecciones de grados 2 a 4 fue del 45,5% en el grupo de R-CHOP y del 42,3% en el grupo de CHOP. Las infecciones micóticas de grados 2 a 4 fueron más frecuentes en el grupo de R-CHOP (4,5% frente al 2,6% en el grupo de CHOP); esta diferencia se debió a una mayor incidencia de candidosis localizadas durante el período de tratamiento. La incidencia de herpes zóster de grados 2 a 4, incluido el herpes zóster oftálmico, fue también mayor en el grupo de R- CHOP (4,5%) que en el grupo de CHOP (1,5%); de los 9 casos registrados en el grupo de R-CHOP, 7 ocurrieron durante la fase de tratamiento. La proporción de pacientes con infecciones de grados 2 a 4 o neutropenia febril fue del 55,4% en el grupo de R-CHOP y del 51,5% en el grupo de CHOP. Neutropenia febril (sin infección simultánea documentada) se notificó únicamente durante el período de tratamiento, en el 20,8% de los pacientes tratados con R-CHOP y el 15,3% de los que habían recibido CHOP. En los pacientes con LLC, la incidencia global de infecciones de grados 3 o 4 durante el tratamiento y los primeros 28 días siguientes a su terminación era comparable en los dos grupos de tratamiento (R-FC: 18%; FC: 17%) y en los pacientes con recidiva/refractarios (R-FC: 19%; FC: 18%). La incidencia de hepatitis B de grados 3 o 4 (reactivación e infección primaria) fue del 2% en el grupo de R-FC frente al 0% en el grupo de FC. Manifestaciones hematológicas: Monoterapia durante 4 semanas: Neutropenia grave (grados 3 o 4) se notificó en el 4,2% de los pacientes; anemia grave, en el 1,1%, y trombocitopenia grave, en el 1,7%. También se han notificado un caso único de anemia aplásica transitoria (aplasia pura de la serie roja) y dos casos de anemia hemolítica tras la administración de MabThera. Terapia de mantenimiento (LNH) hasta 2 años: Leucocitopenia (todos los grados) se produjo en el 21% de los pacientes del grupo de observación frente al 29% en el grupo de MabThera, y neutropenia se notificó en el 12% de los pacientes del grupo de observación y el 23% de los del grupo de MabThera. La incidencia de leucocitopenia (grupo de observación: 2%; grupo de MabThera: 5%) y neutropenia (grupo de observación: 4%; grupo de MabThera: 10%) de grados 3 o 4 fue mayor en el grupo de MabThera que en el de observación. La incidencia de trombocitopenia de grados 3 o 4 (grupo de observación: 1%; grupo de MabThera: < 1%) fue baja. Politerapia (R-CVP en los LNH; R-CHOP en los LDCBG, R-FC en la LLC) Neutropenia grave (grados 3 o 4): La incidencia de neutropenia de grados 3 o 4 en los grupos de MabThera fue mayor que en los grupos de quimioterapia: en el estudio con R-CVP, la incidencia de neutropenia fue del 24% en el grupo de R-CVP frente al 14% en el grupo de CVP. Estos resultados analíticos se notificaron como acontecimientos adversos y dieron lugar a la intervención médica en el 3,1% de los pacientes tratados con R-CVP y el 0,6% de los que recibieron CVP. La incidencia mayor de neutropenia en el grupo de R- CVP no se acompañó de una incidencia mayor de infecciones e infestaciones. En el estudio con R-CHOP, la incidencia de neutropenia grave fue del 97% en el grupo de R- CHOP frente al 88% en el grupo de CHOP. En los pacientes con LLC no tratados previamente, neutropenia de grados 3 o 4 se notificó como acontecimiento adverso en el 30% del grupo de R-FC y el 19% del grupo de FC. En los pacientes con LLC recidivante/refractaria, la incidencia de neutropenia grave de grados 3 o 4 fue ligeramente mayor en el grupo de R-FC (42%) que en el de FC (40%). Leucocitopenia grave (grados 3 o 4): En el estudio con R-CHOP, la incidencia de leucocitopenia grave fue del 88% en el grupo de R-CHOP frente al 79% en el grupo de CHOP. En el tratamiento de primera línea de la LLC, leucocitopenia de grados 3 o 4 se registró en más pacientes tratados con R-FC (23%) que con FC (12%). En los pacientes con LLC recidivante/refractaria, la incidencia global de leucocitopenia de grados 3 o 4 fue comparable en los grupos de tratamiento (R-FC: 4%; FC: 3%). Anemia y trombocitopenia graves (grados 3 o 4): No se observaron diferencias relevantes entre los grupos por lo que respecta a anemia y trombocitopenia de grados 3 o 4. En el estudio con R-CVP, la incidencia de anemia fue del 0,6% en el grupo de R-CVP frente al 1,9% en el grupo de CVP. La incidencia de trombocitopenia fue del 1,2% en el grupo de R-CVP frente al 0% en el grupo de CVP. En el estudio con R-CHOP, la incidencia de anemia fue del 14% en el grupo de R-CHOP frente al 19% en el grupo de CHOP. La incidencia de trombocitopenia fue del 15% en el grupo de R-CHOP frente al 16% en el grupo de CHOP. El tiempo transcurrido hasta la recuperación de todas las alteraciones hematológicas fue similar en ambos grupos. En el estudio sobre tratamiento de primera línea de la LLC se notificó anemia de grados 3 o 4 en el 4% de los pacientes tratados con R-FC frente al 7% de los que recibieron FC, y trombocitopenia de grados 3 o 4 en el 7% del grupo de R-FC frente al 10% en el grupo de FC. En el estudio de la LLC recidivante/refractaria se notificó anemia de grados 3 o 4 en el 12% de los pacientes tratados con R-FC frente al 13% de los que recibieron FC, y trombocitopenia de grados 3 o 4 en el 11% del grupo de R-FC frente al 9% del grupo de FC. Episodios cardiovasculares: Monoterapia durante 4 semanas: En el 18,8% de los pacientes se describieron acontecimientos adversos de tipo cardiovascular durante el período de tratamiento. Los más frecuentes fueron hipotensión e hipertensión. Dos pacientes (0,6%) presentaron arritmia de grados 3 o 4 (incluidas taquicardia ventricular y supraventricular) durante una infusión i.v. de MabThera, y un paciente con antecedentes de infarto de miocardio sufrió angina de pecho, que evolucionó a infarto al cabo de 4 días. Terapia de mantenimiento (LNH) hasta 2 años; La incidencia de trastornos cardíacos de grados 3 o 4 fue comparable en los dos grupos (observación: 4%; MabThera: 5%). Episodios cardíacos graves se notificaron en < 1% de los pacientes del grupo de observación y el 3% de los del grupo de MabThera y consistieron en fibrilación auricular (1%), infarto de miocardio (1%), insuficiencia ventricular izquierda ( < 1%) e isquemia miocárdica ( < 1%). Politerapia (R-CVP en los LNH; R-CHOP en los LDCBG, R-FC en la LLC): En el estudio con R-CVP, la incidencia global de trastornos cardíacos en la población de estudio de la seguridad fue baja (R-CVP: 4%; CVP: 5%), y no hubo diferencias relevantes entre los grupos de tratamiento. En el estudio con R-CHOP, la incidencia de arritmias cardíacas de grados 3 o 4 sobre todo arritmias supraventriculares del tipo de taquicardia supraventricular, aleteo auricular o fibrilación auricular fue superior en el grupo de R-CHOP (14 pacientes, 6,9%) que en el grupo de CHOP (3 pacientes, 1,5%). Todas estas arritmias se presentaron en relación con la infusión de MabThera o estaban asociadas a factores predisponentes, como fiebre, infección, infarto agudo de miocardio o enfermedades respiratorias o cardiovasculares preexistentes (ver Advertencias). No se observaron diferencias entre los grupos de R-CHOP y CHOP en la incidencia de otras reacciones adversas cardíacas de grados 3 o 4, como insuficiencia cardíaca, miocardiopatía o signos de cardiopatía isquémica. En la LLC, la incidencia global de trastornos cardíacos de grados 3 o 4 fue baja tanto en el estudio como tratamiento de primera línea (R-FC: 4%; FC: 3%) como en el estudio de pacientes en recidiva/refractarios (R-FC: 4%; FC: 4%). Concentraciones de IgG: Terapia de mantenimiento (LNH) hasta 2 años: Tras la terapia de inducción, la mediana de las cifras de IgG estaba por debajo del límite inferior de normalidad (LIN) ( < 7 g/l) en ambos grupos: observación y MabThera. En el grupo de observación, la mediana de las cifras de IgG aumentó después por encima del LIN; en cambio, durante el tratamiento con MabThera se mantuvo constante. La proporción de pacientes con concentraciones de IgG por debajo del LIN fue de aproximadamente el 60% en el grupo de MabThera a lo largo de todo el período de tratamiento de 2 años, mientras que en el grupo de observación disminuyó (36% al cabo de 2 años). Manifestaciones neurológicas: Politerapia (R-CVP en los LNH; R-CHOP en los LDCBG, R-FC en la LLC): Durante el período de tratamiento, cuatro pacientes (2%) del grupo de R-CHOP todos ellos con factores de riesgo cardiovascular presentaron accidentes cerebrovasculares de tipo tromboembólico durante el primer ciclo de tratamiento. No se apreciaron diferencias entre ambos grupos en cuanto a incidencia de otros episodios tromboembólicos. En cambio, tres pacientes (1,5%) del grupo de CHOP sufrieron episodios cerebrovasculares, todos ellos durante el período de seguimiento. En la LLC, la incidencia global de trastornos del sistema nervioso de grados 3 o 4 fue baja tanto en el estudio como tratamiento de primera línea (R-FC: 4%; FC: 4%) como en el estudio en pacientes en recidiva/refractarios (R-FC: 3%; FC: 3%). Subpoblaciones: Monoterapia durante 4 semanas: Ancianos (≥ 65 años): La incidencia de RA de cualquier tipo y de RA de grados 3 o 4 fue similar en los ancianos (n = 94) y los pacientes más jóvenes (n = 237) (88,3% frente al 92,0% para cualquier RA y 16,0% frente al 18,1% para RA de grados 3 o 4). Politerapia: Ancianos (≥ 65 años): La incidencia de acontecimientos adversos sanguíneos y linfáticos de grados 3 o 4 fue mayor en los pacientes ancianos (≥ 65 años) que en los más jóvenes entre los pacientes con LLC no tratada previamente o recidivante/refractaria. Enfermedad voluminosa (bulky): La incidencia de RA de grados 3 o 4 fue mayor en los pacientes con enfermedad voluminosa (n = 39) que en los pacientes sin afección voluminosa (n = 195, 25,6% frente al 15,4%). En cambio, la incidencia de RA de cualquier tipo fue similar en ambos grupos (92,3% en el de enfermedad voluminosa frente al 89,2% en el de enfermedad no voluminosa). Retratamiento con monoterapia: El porcentaje de pacientes que notificaron RA de cualquier tipo o RA de grados 3 o 4 tras el retratamiento con nuevas tandas de MabThera (n = 60) fue similar al descrito tras la exposición inicial (n = 203; 95,0% frente al 89,7% para cualquier RA y 13,3% frente al 14,8% para RA de grados 3 o 4). Experiencia de los ensayos clínicos en la artritis reumatoide: A continuación se resume el perfil de seguridad de MabThera en el tratamiento de pacientes con AR moderada o grave. En la población expuesta, más de 3.000 pacientes recibieron como mínimo una tanda de tratamiento y se los mantuvo en seguimiento entre 6 meses y más de 5 años, equivaliendo la exposición global a 7.198 años-paciente; aproximadamente 2.300 pacientes recibieron dos o más tandas de tratamiento durante el período de seguimiento. Las RA enumeradas en la Tabla 3 se basan en los datos de los períodos controlados con placebo de cuatro estudios clínicos multicéntricos de la AR. Los pacientes que recibieron MabThera diferían entre los diferentes estudios: desde pacientes con AR activa en fase precoz no tratados previamente con MTX hasta pacientes con una respuesta inadecuada al MTX (MTX-RI), pasando por pacientes con una respuesta inadecuada a los tratamientos anti-TNF (TNF-RI) (ver Ensayos clínicos / Eficacia). Se administraron 2 veces 1.000 mg o 2 veces 500 mg de MabThera, separadas por un intervalo de dos semanas, además de metotrexato (10-25 mg/semana) (ver Posología en la AR). Las RA enumeradas en la Tabla 3 son las observadas con una frecuencia de al menos el 2%, con una diferencia de al menos el 2% frente al grupo de control, y se presentan independientemente de la dosis. Las frecuencias de la Tabla 3 y la nota correspondiente se definen como muy frecuente (≥ 1/10), frecuente (≥ 1/100 a < 1/10) o poco frecuente (≥ 1/1.000 a < 1/100).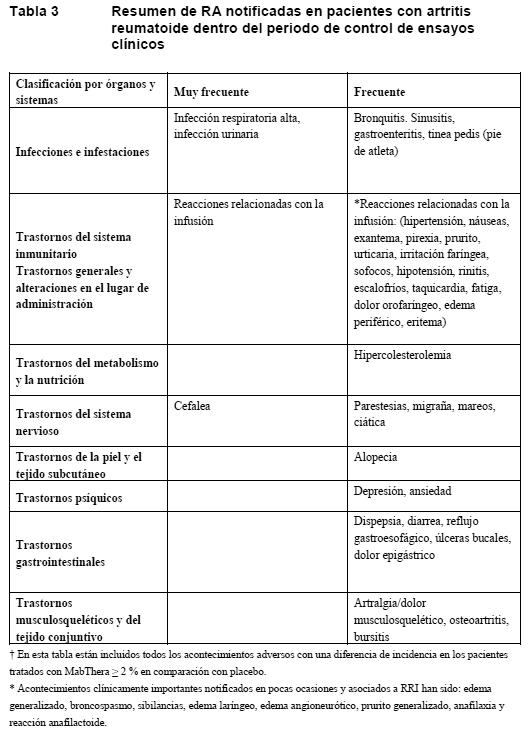
En toda la población expuesta, el perfil de seguridad coincidía con el observado en el período controlado de los estudios clínicos, y no se identificó ninguna nueva RA. Múltiples tandas de tratamiento: El perfil de RA de múltiples tandas de tratamiento es similar al observado tras la primera exposición. El perfil de seguridad mejoró en las tandas siguientes a causa de un descenso de las RRI, las agudizaciones de RA y las infecciones, todas ellas más frecuentes en los 6 primeros meses de tratamiento. Más información sobre reacciones adversas medicamentosas seleccionadas: Reacciones relacionadas con la infusión: En los estudios clínicos, las RA más frecuentes tras la administración de MabThera fueron las reacciones relacionadas con la infusión. De los 3.095 pacientes tratados con MabThera, 1.077 (35%) experimentaron al menos una RRI. La inmensa mayoría de las RRI eran de grados 1 o 2 según los criterios NCI-CTC. En los estudios clínicos, menos del 1% de los pacientes (14 de 3.095) con AR que habían recibido una infusión de MabThera, en cualquier dosis, experimentaron una RRI grave. No hubo ninguna RRI de grados 4 según los criterios NCI-CTC y ninguna muerte por RRI. La proporción de acontecimientos de grados 3 según los criterios NCI-CTC y de RRI que condujeron al abandono disminuyó progresivamente y fueron raros a partir de la tanda 3. En 720/3.095 (23%) pacientes se observaron signos o síntomas indicativos de RRI (náuseas, prurito, fiebre, urticaria/exantema, escalofríos, pirexia, estornudos, edema angioneurótico, irritación faríngea, tos y broncospasmo, con o sin hipotensión o hipertensión asociadas) tras la primera infusión de la primera exposición a MabThera. La premedicación con glucocorticoides por vía i.v. redujo significativamente la incidencia y la gravedad de estas reacciones (ver Advertencias en la AR). Infecciones: La tasa global de infección fue de aproximadamente 97 por 100 años-paciente entre los pacientes tratados con MabThera. Las infecciones fueron predominantemente leves o moderadas y consistieron en la mayoría de los casos en infecciones de las vías respiratorias superiores y del aparato urinario. La tasa de infecciones graves fue de aproximadamente 4 por 100 años-paciente; algunas de ellas fueron letales. Además de las AR recogidas en la Tabla 3, entre los acontecimientos clínicamente graves también se halla la neumonía, con una frecuencia del 1,9%. Neoplasias malignas: La incidencia de tumores malignos tras la exposición a MabThera en los estudios clínicos (0,8 por 100 años-paciente) se halla dentro del intervalo esperado para una población comparable en edad y sexo. Experiencia adquirida en estudios clínicos en las vasculitis asociadas a ANCA (VAA): En el estudio clínico de la VAA, 99 pacientes recibieron tratamiento con MabThera (375 mg/m2, una vez por semana durante 4 semanas) y glucocorticoides (ver Ensayos clínicos/Eficacia). Las reacciones adversas enumeradas en la Tabla 4 fueron todos los acontecimientos adversos registrados con una incidencia ≥ 10% en el grupo de MabThera. Las frecuencias de la Tabla 4 corresponden a la categoría muy frecuente (≥ 1/10).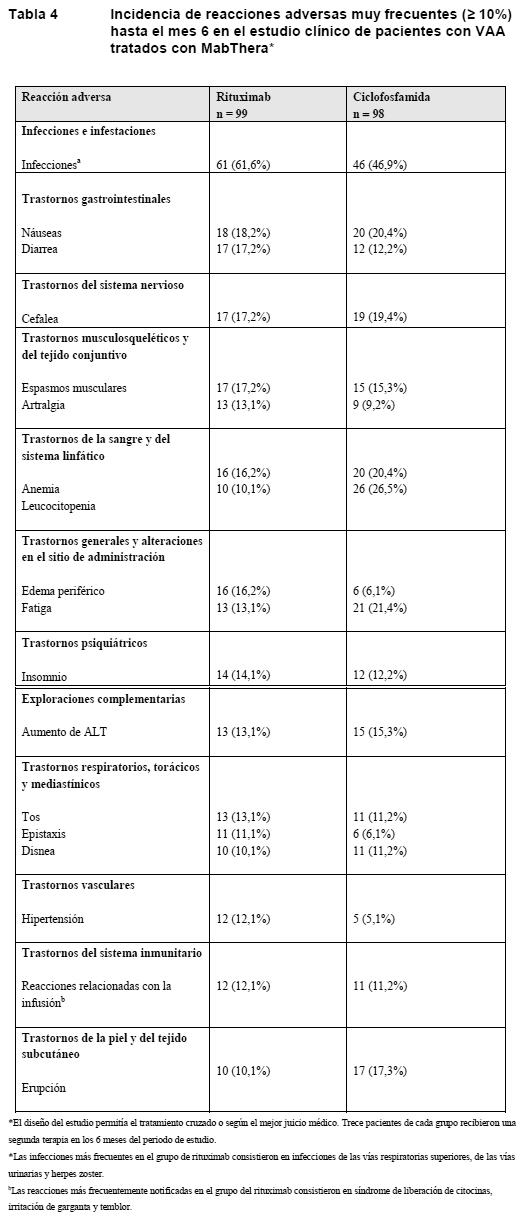
Más información sobre reacciones adversas medicamentosas seleccionadas: Reacciones relacionadas con la infusión: Las reacciones relacionadas con la infusión (RRI) en el estudio de las VAA se definieron como cualquier acontecimiento adverso producido dentro de las 24 horas siguientes a una infusión y considerado por los investigadores como relacionado con la infusión en la población de estudio de la seguridad. Noventa y nueve pacientes recibieron MabThera y el 12% experimentaron al menos una RRI. Todas la RRI fueron de grados 1 o 2 según los criterios CTC. Las RRI más frecuentes consistieron en síndrome de liberación de citocinas, rubefacción, irritación de garganta y temblor. MabThera se administró en combinación con glucocorticoides i.v., que pueden reducir la incidencia y la gravedad de estas reacciones. Infecciones: En los 99 pacientes tratados con MabThera, la tasa global de infecciones fue de aproximadamente 210 por 100 años-paciente (IC del 95%: 173-256). Las infecciones fueron predominantemente leves o moderadas y consistieron en la mayoría de los casos en infecciones de las vías respiratorias superiores, de las vías urinarias y herpes zóster. La tasa de infecciones graves fue de aproximadamente 25 por 100 años-paciente. Neumonía, con una frecuencia del 4%, fue la infección grave más notificada en el grupo de MabThera. Neoplasias malignas: La incidencia de neoplasias malignas en el estudio clínico fue de 2,05 por 100 años- paciente entre los pacientes tratados con MabThera. De acuerdo con los índices de incidencia estandarizados, esta tasa de neoplasias malignas parece similar a las descritas previamente en poblaciones con VAA. Experiencia tras la comercialización: Pacientes con linfoma no hodgkiniano o leucemia linfocítica crónica: Las frecuencias notificadas en este apartado (rara vez, muy rara vez) se basan en estimaciones de la exposición tras la comercialización y en gran medida en datos derivados de notificaciones espontáneas. Tras la comercialización de MabThera se han descrito nuevos casos de reacciones intensas relacionadas con la infusión (ver Advertencias). En el marco del programa de farmacovigilancia continua de MabThera, se han observado las siguientes RA importantes: Aparato cardiovascular: Se han registrado episodios cardíacos graves, consistentes en insuficiencia cardíaca o infarto de miocardio, sobre todo en pacientes con trastornos cardíacos preexistentes o en tratamiento con quimioterápicos cardiotóxicos y, por lo general, relacionados con la infusión. Muy rara vez se ha notificado vasculitis de predominio cutáneo, como la vasculitis leucocitoclástica. Aparato respiratorio: Insuficiencia respiratoria e infiltrados pulmonares asociados a reacciones relacionadas con la infusión (ver Advertencias). Además de trastornos pulmonares asociados con la infusión, se ha descrito enfermedad pulmonar intersticial, en algunos casos letal. Sistemas sanguíneo y linfático: Se han notificado casos de trombocitopenia aguda reversible relacionada con la infusión. Piel y faneras: En raras ocasiones se han notificado reacciones ampollares graves, incluidos algunos casos letales de necrólisis epidérmica tóxica. Sistema nervioso: Se han descrito casos de síndrome de encefalopatía posterior reversible (SEPR) / síndrome de leucoencefalopatía posterior reversible (SLPR). Entre los signos y síntomas se hallan deterioro visual, cefalea, convulsiones y estado mental alterado, con o sin hipertensión asociada. El diagnóstico de SEPR/SLPR requiere la confirmación por imagenología cerebral. Los casos notificados presentaban factores conocidos de riesgo de SEPR/SLPR, incluidos enfermedad subyacente, hipertensión, tratamiento inmunodepresor y/o quimioterapia. En raras ocasiones se ha descrito neuropatía craneal con o sin neuropatía periférica. Se han observado signos y síntomas de neuropatía craneal (por ejemplo: pérdida importante de la vista, el oído u otros sentidos, parálisis facial) en diversos momentos hasta varios meses después de concluido el tratamiento con MabThera. Reacciones generales: En raras ocasiones se han notificado reacciones del tipo de enfermedad del suero. Infecciones e infestaciones: En muy raros casos se ha notificado reactivación de una hepatitis B, la mayoría de las veces en pacientes que recibían rituximab en asociación con quimioterapia antineoplásica (ver Advertencias). Durante el tratamiento con rituximab se han descrito otras infecciones víricas graves unas veces nuevas, otras consistentes en una reactivación o una agudización, algunas de las cuales tuvieron un desenlace letal. La mayoría de los pacientes habían recibido rituximab con quimioterapia o como parte de un trasplante de progenitores hematopoyéticos. Ejemplos de estas graves infecciones víricas son las causadas por virus del herpes (citomegalovirus, CMV), el virus de la varicela-zóster y el virus del herpes simple, el virus JC (leucoencefalopatía multifocal progresiva, PML) y el virus de la hepatitis C. Progresión del sarcoma de Kaposi se ha observado en pacientes expuestos a MabThera con sarcoma de Kaposi preexistente. Estos casos se produjeron en indicaciones no aprobadas y la mayoría de los pacientes eran positivos para el VIH. Tubo digestivo: Se ha observado perforación gastrointestinal, letal en algunos casos, en pacientes con linfoma no hodgkiniano tratados con MabThera en asociación con quimioterapia. Artritis reumatoide: Además de las RA observadas en los estudios clínicos de la AR con rituximab (ver Ensayos clínicos, Experiencia de los ensayos clínicos en la artritis reumatoide), se han descrito leucoencefalopatía multifocal progresiva y reacciones del tipo de enfermedad del suero tras la comercialización. Alteraciones analíticas: Linfomas no hodgkinianos: Sistemas sanguíneo y linfático: Neutropenia: En raras ocasiones se ha instaurado neutropenia más de cuatro semanas después de la última infusión de MabThera. Tras la comercialización: En estudios de rituximab en pacientes con enfermedad de Waldenström se han observado aumentos transitorios de las concentraciones séricas de IgM tras el inicio del tratamiento, lo que puede estar asociado a hiperviscosidad y síntomas relacionados. Por lo general, el incremento transitorio de IgM disminuyó al menos a las cifras basales en el espacio de 4 meses.
Advertencias.
Pacientes con linfoma no hodgkiniano o leucemia linfocítica crónica: Reacciones relacionadas con la infusión: La administración de MabThera en infusión comporta reacciones que pueden estar relacionadas con la liberación de citocinas u otros mediadores químicos. Clínicamente, una reacción grave relacionada con la infusión puede ser indistinguible de una reacción alérgica o del síndrome de liberación de citocinas. En el uso tras la comercialización se han descrito reacciones graves relacionadas con la infusión que tuvieron un desenlace fatal. Las reacciones graves relacionadas con la infusión se presentaron habitualmente al cabo de 30 minutos a 2 horas de iniciada la primera infusión de MabThera, se caracterizaban por episodios pulmonares e incluían en algunos casos lisis tumoral aguda y ciertas características del síndrome de lisis tumoral, además de fiebre, escalofríos, hipotensión, urticaria, angioedema y otros síntomas (ver Advertencias y Reacciones a
Interacciones.
Hasta el presente son limitados los datos sobre la posibilidad de interacción farmacológica con MabThera. En los pacientes con LLC, la coadministración con MabThera no parecía tener ningún efecto en la farmacocinética de la fludarabina y la ciclofosfamida; por otro lado, no se observó ningún efecto de la fludarabina ni de la ciclofosfamida en la farmacocinética de MabThera. La coadministración de MTX no tenía ningún efecto en la farmacocinética de MabThera en los pacientes con AR. Los pacientes con anticuerpos humanos antimurinos (HAMA) o antiquiméricos (HACA) pueden experimentar reacciones alérgicas si reciben otros anticuerpos monoclonales terapéuticos o para diagnóstico. En el programa de estudios clínicos de la AR, 373 pacientes tratados con MabThera recibieron tratamiento ulterior con otros FAME; de ellos, 240 recibieron un FAME biológico. En estos pacientes, la tasa de infección grave durante el tratamiento con MabThera (antes del FAME biológico) fue de 6,1 por 100 años-paciente, frente a 4,9 por 100 años-paciente tras el tratamiento con el FAME biológico. Uso en poblaciones especiales: Embarazo: Se sabe que las inmunoglobulinas IgG atraviesan la barrera placentaria. Los estudios de toxicidad en el desarrollo realizados con macacos de Java (Macaca fascicularis) no han revelado ningún indicio de embriotoxicidad en el útero. En las crías recién nacidas de madres expuestas a MabThera se observó depleción de poblaciones de células B durante la fase posnatal. No se han estudiado en ensayos clínicos las cifras de células B en neonatos humanos tras la exposición materna a MabThera. Aunque no hay datos adecuados y bien controlados de estudios en mujeres embarazadas, se han notificado depleción transitoria de células B y linfocitopenia en algunos lactantes de madres expuestas al rituximab durante el embarazo. Por ello, MabThera no debe administrarse a mujeres embarazadas, salvo que los beneficios esperados sean mayores que el riesgo. Las mujeres en edad fértil deben aplicar métodos anticonceptivos eficaces durante el tratamiento con MabThera y los 12 meses siguientes. Lactancia: No se sabe si el rituximab pasa a la leche materna. Teniendo en cuenta, sin embargo, que las IgG de la madre pasan a la leche materna, no debe administrarse MabThera a madres lactantes.
Conservación.
Este medicamento no debe utilizarse después de la fecha de caducidad, indicada con EXP en el envase. Consérvense los viales a 2-8°C (en un refrigerador). Manténgase el envase en el embalaje externo para protegerlo de la luz. Desde un punto de vista microbiológico, la solución para infusión preparada debe utilizarse inmediatamente. Si no se utiliza inmediatamente, la duración y las condiciones de conservación del producto antes de su utilización son de la responsabilidad del usuario. Habitualmente, no debería sobrepasar las 24 horas a 2-8°C, salvo que la dilución se haya realizado en condiciones asépticas controladas y validadas. Instrucciones especiales de uso, manipulación y eliminación: Extráigase la cantidad necesaria de MabThera en condiciones asépticas y dilúyase hasta una concentración calculada de rituximab de 1-4 mg/ml en una bolsa de infusión con suero fisiológico al 0,9%, estéril y apirógeno, o una solución acuosa de glucosa al 5%. Para mezclar la solución, inviértase lentamente la bolsa con objeto de evitar la formación de espuma. Los medicamentos parenterales deben inspeccionarse visualmente antes de la administración para detectar eventuales partículas en suspensión o descoloración. Las soluciones de MabThera preparadas para la infusión permanecen física y químicamente estables durante 24 horas a 2-8°C más otras 12 horas a temperatura ambiente. Incompatibilidades: No se han descrito incompatibilidades entre MabThera y las bolsas o equipos de infusión de cloruro de polivinilo o polietileno. Eliminación de medicamentos no utilizados/caducados: La emisión de productos farmacéuticos al medio ambiente debe reducirse a un mínimo. Los medicamentos no deben eliminarse a través de las aguas residuales, y su eliminación con los residuos domésticos también debe evitarse. Utilice los sistemas de recogida establecidos si los hay en su localidad.
Sobredosificación.
No se ha descrito ningún caso de sobredosis en los estudios clínicos. No se han ensayado dosis únicas superiores a 1.000 mg en estudios clínicos controlados. La dosis más alta ensayada hasta la fecha ha sido de 5 g en pacientes con leucemia linfocítica crónica. No se conoce ninguna otra señal de posible riesgo. En caso de sobredosis, debe suspenderse o reducirse inmediatamente la infusión y vigilar estrechamente al paciente. Se debe considerar la idoneidad de controlar regularmente el hemograma completo y tener en cuenta el mayor riesgo de infección mientras dura la depleción de células B.
Presentación.
Viales de 10 ml (10 mg/ml): envase con 2. Viales de 50 ml (10 mg/ml): envase con 1.


 Cambiar de país
Cambiar de país