KALETRA
ABBOTT
Antiviral.
Descripción.
Lopinavir/ritonavir es una co-formulación de lopinavir y ritonavir. Lopinavir es un inhibidor de las proteasas VIH-1 y VIH-2. Al ser una co-formulación en lopinavir/ritonavir, ritonavir inhibe el CYP3A (citocromo P450 3A) que media en el metabolismo de lopinavir, por lo tanto, provee un aumento de los niveles plasmáticos de lopinavir. El lopinavir es un polvo de color blanco a levemente tostado. Es libremente soluble en metanol y etanol, soluble en isopropanol y prácticamente insoluble en agua. Lopinavir es químicamente designado como un [1S-[1R*,(R*),3R*,4R*]]-N-[4-[(2,6-dimetilfenoxi)acetil]amino]-3-hidroxi-5-fenil-1-(fenilmetil)pentil]tetrahidro-alpha-(1-metiletil)-2-oxo-1 (2H)-pirimidineacetamida. Su fórmula molecular es C37H48 N4 O5 y su peso molecular es de 628,80. Lopinavir tiene la siguiente fórmula estructural: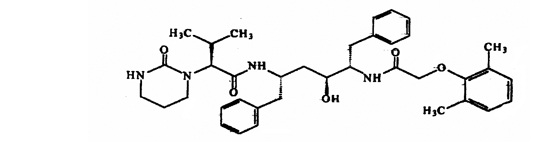
Ritonavir químicamente se designa como 10-Hydroxy-2-methyl-5-(1-methylethyl)-1-[2-(1-methylethyl)-4-thiazolyl]-3,6-dioxo-8,11bis(phenylmethyl)-2,4,7,12-tetraazatridecan-13-oic acid, 5-thiazolylmethyl ester, [5S-(5R*,8R*,10R*,11R*)]. Su fórmula molecular es C37 H48 N6 O5 S2 y su peso molecular es de 720,95. Ritonavir tiene la siguiente estructura química: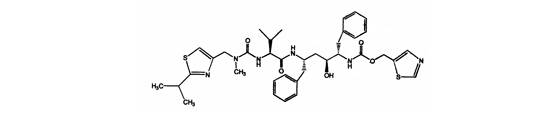
Los comprimidos recubiertos de Lopinavir/ritonavir están disponibles para la administración oral en dosis de 200 mg de lopinavir y 50 mg de ritonavir con los siguientes ingredientes inactivos: copovidona, laurato de sorbitan, dióxido de silicio coloidal, estearil fumarato de sodio, hipromelosa 2910, dióxido de titanio, macrogol 400, hidroxipropilcelulosa, hipromelosa 2910, talco, dióxido de silicio coloidal anhidro, macrogol 3350, polisorbato 80, óxido de hierro amarillo. También están disponibles en dosis de 100 mg de lopinavir y 25 mg de ritonavir con los siguientes ingredientes inactivos: copovidona, laurato de sorbitan, dióxido de silicio coloidal, estearil fumarato de sodio, alcohol polivinílico, dióxido de titanio, talco, macrogol 3350, óxido de hierro amarillo.
Farmacología.
Microbiología: Mecanismo de acción: El lopinavir, un inhibidor de las proteasas del VIH-1 y VIH-2, impide el clivaje de la poliproteína gag-pol, dando como resultado la producción de virus inmaduros no infecciosos. Actividad antiviral in vitro: Se evaluó la actividad antiviral in vitro del lopinavir contra cepas de VIH de laboratorio y aislados clínicos de VIH en líneas celulares linfoblásticas agudamente infectadas y en linfocitos de sangre periférica, respectivamente. En ausencia de suero humano, la concentración efectiva media 50% (EC50) de lopinavir contra cinco diferentes cepas de laboratorio de VIH-1 varió de 10 a 27 nM (0,006 a 0,017 mcg/mL, 1 mcg/mL igual a 1,6 microM) y varió de 4 a 11 nM (0,003 a 0,007 mcg/mL) contra varios aislados clínicos de VIH-1 (n igual a 6). En presencia de 50% de suero humano, la media de CE50 de lopinavir contra estas cinco cepas de laboratorio varió de 65 a 289 nM (0,04 a 0,18 mcg/mL), representando una atenuación de 7 a 11 veces. Estudios sobre actividad de combinación de droga con lopinavir y otros inhibidores de proteasa o inhibidores de transcriptasa reversa no se han completado. Resistencia: Se han seleccionado aislados de VIH-1 con susceptibilidad reducida a lopinavir in vitro. La presencia de ritonavir no parece influenciar la selección de los virus resistentes a lopinavir in-vitro. La selección de resistencia a lopinavir/ritonavir en tratamiento antirretroviral en pacientes naive no ha sido caracterizada. En un estudio fase III con 653 pacientes naive a tratamiento antirretroviral (estudio 863), se analizaron aislados virales plasmáticos de cada paciente en tratamiento con un VIH plasmático mayor que 400 copias/mL en la semana 24, 32, 40 y/o 48. No se observó ninguna evidencia de resistencia genotípica o fenotípica a lopinavir/ritonavir en 37 pacientes evaluables tratados con lopinavir/ritonavir (0%). La evidencia de resistencia genotípica a nelfinavir, definida como la presencia de la mutación D30N y/o L90M en la proteasa del VIH, se observó en 25/76 (33%) de pacientes evaluables tratados con nelfinavir. La selección de la resistencia a lopinavir/ritonavir en pacientes pediátricos naive a tratamiento antirretroviral (estudio 940) parece ser consistente con lo observado en los pacientes adultos (estudio 863). Resistencia a lopinavir/ritonavir se ha observado que aparece en pacientes tratados con otros inhibidores de proteasa previo a la terapia con lopinavir/ritonavir. En estudios Fase II de 227 pacientes naive a tratamiento antirretroviral y que han utilizado inhibidor de proteasa, aislados de 4 de 23 pacientes con RNA viral cuantificable (mayor de 400 copias/mL) después del tratamiento con lopinavir/ritonavir por 12 a 100 semanas mostraron susceptibilidad significativamente reducida a lopinavir comparada con los aislados virales correspondientes al estado basal. Tres de estos pacientes habían recibido previamente tratamiento con un solo inhibidor de proteasa (nelfinavir, indinavir o saquinavir) y un paciente había recibido tratamiento con múltiples inhibidores de proteasa (indinavir, saquinavir y ritonavir). Cuatro de estos pacientes tenían al menos cuatro mutaciones asociadas con resistencia al inhibidor de proteasa inmediatamente anterior a la terapia con lopinavir/ritonavir. Siguiente al rebote viral, los aislados de todos estos pacientes contenían mutaciones adicionales, algunas de las cuales se reconocen estar asociadas con resistencia al inhibidor de proteasa. Sin embargo, en este momento existen datos insuficientes para identificar los patrones de mutación asociados a lopinavir en aislados de pacientes en terapia con lopinavir/ritonavir. La evaluación de estos patrones mutacionales está bajo estudio. Resistencia-cruzada: Estudios preclínicos: Se han observado grados variados de resistencia cruzada entre los inhibidores de proteasa. La actividad in-vitro de lopinavir contra los aislados clínicos de pacientes previamente tratados con un solo inhibidor de proteasa fue determinado. Aislados que mostraron más de 4-veces de susceptibilidad reducida a nelfinavir (n=13) y saquinavir (n=4), mostraron menos de 4-veces de susceptibilidad reducida a lopinavir. Aislados con más de 4-veces desusceptibilidad reducida a indinavir (n=16) y ritonavir (n=3) mostraron una media de 5,7 y 8,3-veces de susceptibilidad reducida a lopinavir, respectivamente. Aislados de pacientes previamente tratados con dos o más inhibidores de proteasa mostraron mayores reducciones en la susceptibilidad a lopinavir, como se describe en la sección clínica más abajo (ver Estudios Clínicos: Actividad Antiviral de lopinavir/ritonavir en Pacientes Con Terapia Previa de Inhibidor de Proteasa). Resistencia Cruzada Durante la Terapia con lopinavir/ritonavir: Se dispone de poca información sobre la resistencia cruzada de virus seleccionados durante la terapia con lopinavir/ritonavir. Aislados de cuatro pacientes previamente tratados con uno o más inhibidores de proteasa que desarrollaron resistencia fenotípica aumentada a lopinavir durante la terapia con lopinavir/ritonavir permanecieron ya sea con resistencia cruzada o desarrollaron resistencia cruzada a ritonavir, indinavir y nelfinavir. Todos los rebotes virales ya sea permanecieron totalmente sensibles o demostraron susceptibilidad modestamente reducida a amprenavir (hasta 8,5-veces concurrente con 99-veces de resistencia a lopinavir). El rebote de aislados de dos sujetos sin tratamiento previo con saquinavir permanecieron totalmente sensibles a saquinavir. Correlación genotípica de la respuesta virológica disminuida en pacientes que han utilizado antirretrovirales que inician un régimen de combinación basado en lopinavir/ritonavir: Se ha mostrado que la respuesta virológica a lopinavir/ritonavir se afecta por la presencia de tres o más de las siguientes substituciones de aminoácidos en la proteasa en el estado basal: L10F/I/R/V, K20M/N/R, L24I, L33F, M36I, I47V, G48V, I54L/T/V, 82A/C/F/S/T, e I84V. La Tabla 7 muestra la respuesta virológica a las 48 semanas (ARN VIH < 400 copias/mL) de acuerdo al número de mutaciones de resistencia de los inhibidores de proteasa mencionados anteriormente en el estado basal en los estudios 888 y 765 y estudio 957 (ver a continuación).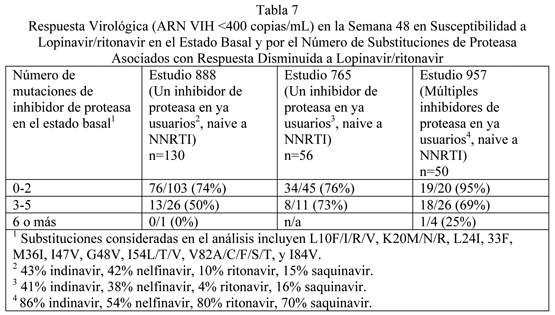
La Tabla 8 muestra la respuesta virológica a 48 semanas (RNA VIH-1 < 50 copias/mL) en el estudio 802 de acuerdo al número de mutaciones de resistencia asociadas a lopinavir listadas en la Tabla 7 presentes en el estado basal. Existen datos insuficientes para apoyar la administración una vez al día de lopinavir/ritonavir para pacientes adultos con tres o más mutaciones asociadas a lopinavir.
Estudios Clínicos: Actividad Antiviral de lopinavir/ritonavir en Pacientes con Tratamiento Previo con Inhibidor de la Proteasa: Se ha examinado la relevancia clínica de la susceptibilidad reducida in vitro a lopinavir mediante evaluación de la respuesta virológica al tratamiento con lopinavir/ritonavir, con respecto al genotipo y fenotipo viral basal, en 56 pacientes naive a NNRTI con ARN VIH > 1000 copias/mL a pesar del tratamiento previo con al menos dos inhibidores de proteasa seleccionados de nelfinavir, indinavir, saquinavir y ritonavir (Estudio M98-957). En este estudio, los pacientes se randomizaron inicialmente para recibir una de dos dosis de lopinavir/ritonavir en combinación con efavirenz e inhibidores nucleósidos de transcriptasa reversa. Los valores de CE50 de lopinavir contra los 56 aislados virales basales varió de 0.5 a 96 veces mayores que la CE50 contra el VIH de tipo salvaje. Cincuenta y cinco por ciento (31/56) de estos aislados basales mostraron una susceptibilidad reducida de más de 4-veces a lopinavir. Estos 31 aislados tuvieron una reducción media de la susceptibilidad a lopinavir de 27,9 veces. Después de 48 semanas de tratamiento con lopinavir/ritonavir, efavirenz e inhibidores nucleósidos de la transcriptasa reversa, se observaron ARN VIH plasmático menores o iguales a 400 copias /mL en 93% (25/27), 73% (11/15), y 25% (2/8) de los pacientes con menos de o igual a 10 veces, mayor que 10 y menos que 40 veces, y mayor que o igual a 40 veces la susceptibilidad reducida a lopinavir en el estado basal, respectivamente. La susceptibilidad a lopinavir se determinó por tecnología fenotípica recombinante realizada por Virologic; el genotipo también lo realizó Virologic. RNA VIH plasmático menor que o igual a 50 copias /mL se observó en 81% (22/27), 60% (9/15) y 25% (2/8) en los grupos de pacientes arriba mencionados, respectivamente. Existen datos insuficientes en estos momentos para identificar los patrones mutacionales asociados a lopinavir en aislados de pacientes en terapia con lopinavir/ritonavir. Se necesitan estudios adicionales para evaluar la asociación entre patrones mutacionales específicos y tasas de respuesta virológica. Farmacocinética: Se han evaluado las propiedades farmacocinéticas de lopinavir coadministrado con ritonavir en voluntarios adultos sanos y en pacientes infectados con VIH no se observaron diferencias significativas entre ambos grupos. El lopinavir se metaboliza fundamentalmente completamente por CYP3A. El ritonavir inhibe el metabolismo de lopinavir, por consiguiente aumentan los niveles plasmáticos de lopinavir. A través de los estudios clínicos, la administración de 400/100 mg de lopinavir/ritonavir dos veces por día produjo concentraciones medias plasmáticas estado estable de lopinavir 15 a 20 veces superiores a las de ritonavir en pacientes infectados con VIH. Las concentraciones plasmáticas de ritonavir son menores que 7% de aquellas obtenidas después de dosis de 600 mg de ritonavir dos veces por día. La CE50 antiviral in vitro de lopinavir es aproximadamente 10 veces menor que la de ritonavir. Por lo tanto, la actividad antiviral de Kaletra se debe a lopinavir. La Figura 1 muestra las concentraciones plasmáticas medias estado estable de lopinavir y ritonavir después de la administración de 400/100 mg dos veces al día con alimentos durante tres semanas de un estudio farmacocinético en sujetos adultos infectados con VIH (n=19).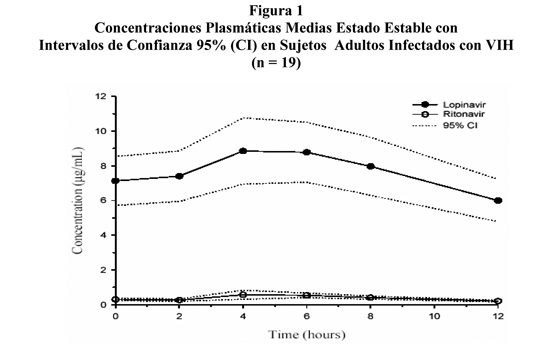
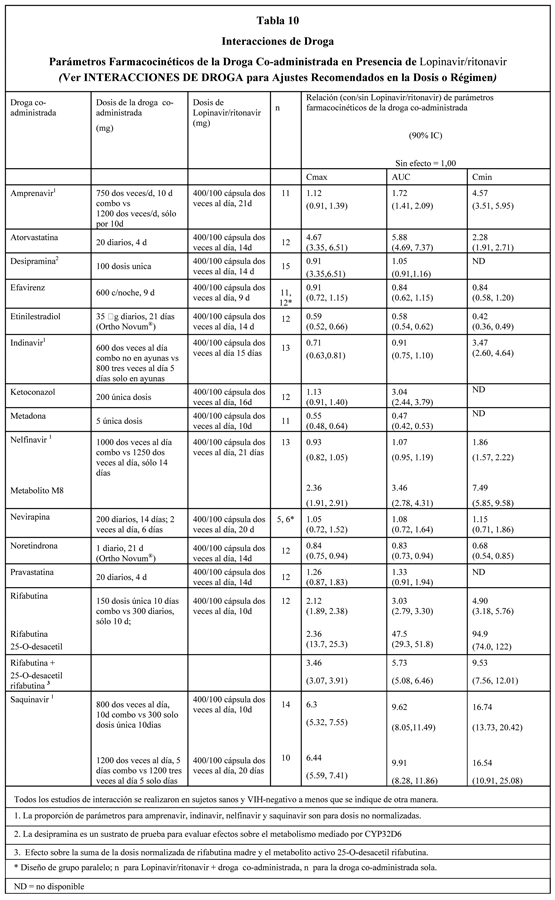
Las concentraciones plasmáticas de lopinavir y ritonavir después de la administración de dos comprimidos de 200/50 mg son equivalentes a tres cápsulas de 133/33 mg administradas bajo condiciones de alimentación con menos variabilidad farmacocinética. Absorción: En un estudio farmacocinético en sujetos VIH-Positivos (n=18), con dosis múltiples de lopinavir/ritonavir 400/100 mg dos veces al día administradas con el alimento por tres semanas, produjo una media ± DS del peak de la concentración plasmática de lopinavir (Cmáx) de 12.3 ± 5.4 mcg/mL, ocurriendo aproximadamente cuatro horas después de la administración. La media de la concentración mínima estado estable previo a la dosis de la mañana fue 8.1 ± 5.7 mcg/mL y la concentración mínima dentro de un intervalo de dosificación fue 5.6 ± 4.5 mcg/mL. El AUC de Lopinavir sobre un intervalo de dosificación de 12 horas promedió 113.2 ± 60.5 mcg•h/mL. La biodisponibilidad absoluta de lopinavir co-formulado con ritonavir en seres humanos no se ha establecido. Efectos de las Comidas sobre la Absorción Oral: La administración de una sola dosis de 400/100 mg de lopinavir/ritonavir comprimidos bajo condiciones de alimentación (alto grado en grasas, 872 kcal, 56% de grasa) comparada al estado de ayunas no se asoció con cambios significativos en Cmáx y AUCinf, por lo tanto, lopinavir/ritonavir comprimidos, se puede tomar con o sin alimentos. lopinavir/ritonavir comprimidos también ha mostrado menos variabilidad farmacocinética bajo todas las condiciones de alimentación comparado con lopinavir/ritonavir cápsula. Distribución: En estado estable, lopinavir se une aproximadamente 98 a 99% a las proteínas plasmáticas. Lopinavir se une tanto a la glucoproteína ácida alfa-1 (GAA) como a la albúmina, sin embargo, tiene una mayor afinidad por la GAA. En estado estable, la unión de lopinavir a las proteínas se mantiene constante sobre el rango de concentraciones observadas después de la administración de 400/100 mg de lopinavir/ritonavir dos veces por día y es similar entre voluntarios sanos y pacientes VIH-positivos. Metabolismo: Experimentos in vitro con microsomas hepáticos humanos indican que lopinavir principalmente experimenta un metabolismo oxidativo. El lopinavir es extensamente metabolizado por el sistema hepático del citocromo P450, casi exclusivamente por la isoenzima CYP3A. El ritonavir es un potente inhibidor de la CYP3A, el cual inhibe el metabolismo de lopinavir y, en consecuencia, aumenta los niveles plasmáticos de lopinavir. Un estudio con lopinavir C14 en seres humanos mostró que 89% de la radioactividad plasmática después de una dosis única de 400/100 mg de lopinavir/ritonavir se debió a la droga madre. Se han identificado por lo menos 13 metabolitos oxidativos de lopinavir en seres humanos. El ritonavir ha mostrado inducir enzimas metabólicas, provocando la inducción de su propio metabolismo. Con dosis múltiples, las concentraciones predosis de lopinavir disminuyen con el tiempo, estabilizándose después de aproximadamente 10 a 16 días. Eliminación: Después de una dosis de 400/100 mg de lopinavir C14/ ritonavir, aproximadamente 10,4 ± 2,3% y 82,6 ± 2,5% de una dosis administrada de lopinavir C14 se puede encontrar en la orina y heces, respectivamente, después de 8 días. Alrededor de 2,2% y 19,8% de la dosis administrada de lopinavir se encuentra sin cambios en la orina y heces, respectivamente. Después de dosis múltiples, menos del 3% de la dosis de lopinavir se excreta en sin cambios en la orina. El clearance oral aparente (CL/F) de lopinavir es 5.98 ± 5.75 L/hr (media ± DS, N=19). Dosificación una vez al día: La farmacocinética de una vez al día de lopinavir/ritonavir se ha evaluada en sujetos infectados con VIH naive a tratamiento antirretroviral. lopinavir/ritonavir 800/200 mg se administró en combinación con emtricitabina 200 mg y tenofovir DF 300 mg como parte de un régimen una vez al día. Múltiples dosis de lopinavir/ritonavir 800/200 mg una vez al día por 2 semanas sin restricción de alimentos (n = 16) produjo una media ± DS de concentración plasmática peak de lopinavir (Cmáx) de 14.8 ± 3.5 mg/mL, el que ocurrió aproximadamente 6 horas después de la administración. La media estado estable de la concentración mínima de lopinavir previo a la dosis de la mañana fue 5.5 ± 5.4 mg/mL y la concentración mínima dentro de un intervalo de dosis fue 3.2 ± 3.4 mg/mL. El AUC de Lopinavir sobre un intervalo de dosis de 24 horas promedió 206.5 ± 89.7 mg*h/mL. Efectos en el Electrocardiograma: Se han reportado casos post-marketing de prolongación del intervalo QT y torsades de pointes, aunque no se ha establecido una causalidad respecto a Kaletra. Evite el uso de Kaletra en pacientes con prolongación del intervalo QT congénito. El intervalo QTcF se evaluó en un estudio randomizado, placebo y activo (moxifloxacino 400 mg una vez al día) controlado, cruzado en 39 adultos sanos, con 10 mediciones sobre 12 horas en el Día 3. Las diferencias máximas medias (límite superior de confianza 95%) en QTcF del placebo eran 3.6 (6.3) mseg y 13.1 (15.8) mseg para lopinavir/ritonavir 400/100 mg dos veces al día y supraterapéutico 800/200 mg dos veces al día, respectivamente. Los dos regímenes produjeron exposiciones en el Día 3 que fueron aproximadamente 1.5 y 3 veces mayores que aquellas observadas con la dosis recomendada de lopinavir/ritonavir una vez al día o dos veces al día en estado estable. Ningún sujeto presentó un aumento en QTcF de ≥60 mseg desde el estado basal o un intervalo QTcF que excediera el umbral potencialmente clínicamente relevante de 500 mseg. Una modesta prolongación del intervalo PR también se observó en sujetos que recibían lopinavir/ritonavir en el mismo estudio en el Día 3. El intervalo PR máximo fue 286 mseg y no se observó bloqueo cardíaco de segundo o tercer grado (ver Advertencias). Poblaciones especiales: Sexo, Raza y Edad: No se ha estudiado la farmacocinética de lopinavir en pacientes geriátricos. En pacientes adultos no se han observado diferencias farmacocinéticas relacionadas con el sexo. No se han identificado diferencias farmacocinéticas debidas a la raza. Pacientes Pediátricos: Se ha estudiado la farmacocinética de lopinavir/ritonavir en dosis de 300/75 mg/m2 y 230/57,5 mg/m2 administradas dos veces por día en un total de 53 pacientes pediátricos con edades comprendidas entre 6 meses a 12 años. El régimen de 230/57,5 mg/m2 dos veces al día sin nevirapina y el régimen de 300/75 mg/m2 dos veces al día con nevirapina proporcionaron concentraciones plasmáticas de lopinavir similares a aquellas obtenidas en pacientes adultos que recibieron el régimen de 400/100 mg dos veces al día (sin nevirapina). lopinavir/ritonavir una vez al día no se ha evaluado en pacientes pediátricos. La media de AUC, Cmáx y Cmín de lopinavir en estado estable fueron 72,6 ± 31,1 mcg·h/mL, 8,2 ± 2,9 y 3,4 ± 2,1 mcg/mL, respectivamente, después de la administración de 230/57,5 mg/m2 de lopinavir/ritonavir dos veces por día sin nevirapina (n=12) y fueron 85,8 ± 36,9 mcg·h/mL, 10,0 ± 3,3 y 3,6 ± 3,5 mcg/mL, respectivamente luego de 300/75 mg/m2 dos veces por día con nevirapina (n=12). El régimen de nevirapina fue 7 mg/kg dos veces por día (en niños de seis meses a 8 años) o 4 mg/kg dos veces por día (en niños mayores de ocho años). Insuficiencia Renal: No se ha estudiado la farmacocinética de lopinavir en pacientes con insuficiencia renal; sin embargo, debido a que el clearance renal de lopinavir es mínimo, no se espera que el clearance corporal total disminuya en pacientes con insuficiencia renal. Insuficiencia Hepática: Lopinavir se metaboliza y se elimina principalmente por el hígado. La múltiple dosificación de lopinavir/ritonavir 400/100 mg dos veces al día en pacientes co-infectados con VIH y HCV con daño hepático leve a moderado produjo un aumento del 30% en el AUC de lopinavir y aumentó 20% en Cmáx comparado a los sujetos infectados con VIH con función hepática normal. Además, la unión a las proteínas plasmáticas de lopinavir fue menor tanto en el daño hepático leve como moderado comparada con los controles (99.09 vs. 99.31% respectivamente). lopinavir/ritonavir no se ha estudiado en pacientes con daño hepático severo (ver Advertencias). Interacciones Droga-Droga: (ver también Contraindicaciones, Advertencias e Interacciones de droga). lopinavir/ritonavir es un inhibidor in vitro de la isoforma CYP3A del citocromo P450. La co-administración de lopinavir/ritonavir y otras drogas principalmente metabolizadas por CYP3A puede provocar un aumento de las concentraciones plasmáticas de otras drogas, las cuales podrían aumentar o prolongar sus efectos terapéuticos y adversos (ver Contraindicaciones). lopinavir/ritonavir no inhibe las isoformas CYP2D6, CYP2C9, CYP2C19, CYP2E1, CYP2B6 o CYP1A2 en concentraciones clínicamente relevantes. lopinavir/ritonavir ha mostrado inducir in vivo su propio metabolismo y aumentar la biotransformación de algunas drogas metabolizadas por las enzimas del citocromo P450 y por glucuronización. lopinavir/ritonavir es metabolizado por el CYP3A. Las drogas que inducen actividad CYP3A se esperaría que aumenten el clearance de lopinavir, resultando en concentraciones plasmáticas de lopinavir más bajas. Aunque no se ha observado con ketoconazol concomitante, la co-administración de lopinavir/ritonavir y otras drogas que inhiben la CYP3A pueden aumentar las concentraciones plasmáticas de lopinavir. Se llevaron a cabo estudios de interacciones de droga entre lopinavir/ritonavir y otras drogas probables a ser co-administrados y algunas drogas comúnmente usadas como controles para interacciones farmacocinéticas. Los efectos de la co-administración de lopinavir/ritonavir sobre el AUC, Cmáx y Cmín se resumen en la Tabla 9 (efecto de otras drogas sobre lopinavir) y la Tabla 10 (efectos de lopinavir/ritonavir sobre otras drogas). Los efectos de otras drogas sobre ritonavir no se muestran ya que generalmente se correlacionan con aquellos observados con lopinavir (si las concentraciones de lopinavir se disminuyen, también se disminuyen las concentraciones de ritonavir) a menos que de lo contrario indicado al pie de la tabla. Para información sobre recomendaciones clínicas, ver Interacciones de droga.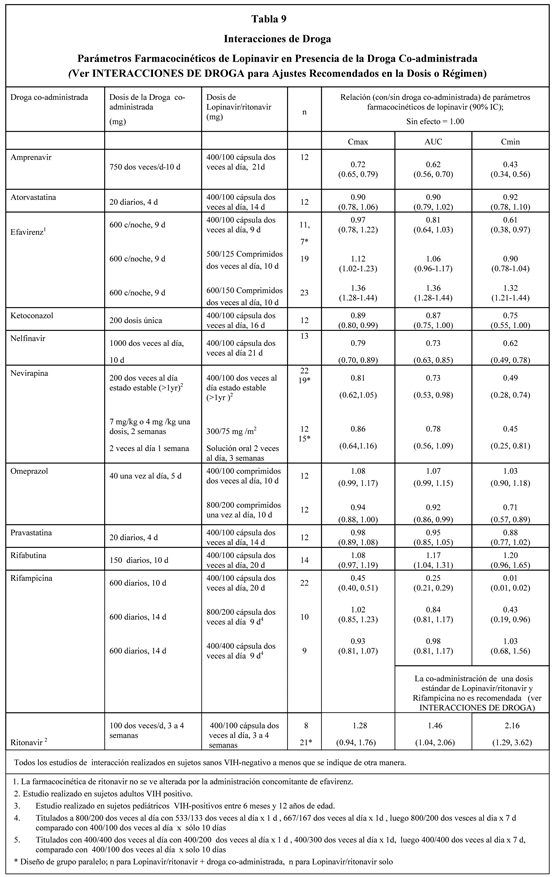
Datos de seguridad pre-clínicos: Toxicidad aguda, semiaguda y crónica: Estudios de toxicidad con dosis repetidas en roedores y perros identificados los órganos objetivo principales como el hígado, riñón, tiroides, bazo, y glóbulos rojos circulantes. Los cambios hepáticos indicaron inflamación celular con degeneración focal. Dado que la exposición produjo estos cambios que fueron comparables con la exposición clínica en humanos, las dosis en animales fueron sobre 6 veces la dosis clínica recomendada. La degeneración tubular renal leve se limitó a ratones expuestos con al menos dos veces la exposición recomendada en humanos; en ratas y perros el riñón no fue afectado. La tiroxina sérica reducida provocó un aumento de TSH con una hipertrofia celular folicular resultante en las glándulas tiroideas de las ratas. Estos cambios fueron reversibles con el retiro de la sustancia activa y no se apreciaron en ratones y perros. Se observó Coombs-negativo, poiquilocitosis y anisocitosis en ratas, pero no en ratones ni perros. Se observó bazo de mayor tamaño con histiocitosis en ratas pero no en otras especies. El colesterol sérico se elevó en roedores pero no en perros, mientras que los triglicéridos se elevaron sólo en ratones. Carcinogénesis y Mutagénesis: Los estudios de carcinogenicidad a largo plazo de lopinavir/ritonavir en ratones revelaron una inducción mitogénica no-genotóxica de tumores hepáticos, considerada generalmente de poca relevancia en el riesgo humano. Los estudios de carcinogenicidad en ratas no revelaron hallazgos tumorigénicos. Lopinavir no se encontró ser mutagénico o clastogénico en una batería de ensayos in vitro incluyendo el ensayo de mutación reversa bacteriano de Ames, el ensayo de linfoma de ratón, y ensayos de aberración cromosómica en linfocitos humanos. Lopinavir/ritonavir no se encontró ser mutagénico o clastogénico en ensayos in vivo usando el ensayo de micronúcleos de ratón. Descripción de estudios clínicos: Pacientes sin Terapia Antiretroviral Previa: Estudio M98-863: lopinavir/ritonavir cápsulas dos veces al día + estavudina + lamivudina comparada con nelfinavir TID + estavudina + lamivudina: El estudio M98-863 era un estudio multicéntrico, aleatorio, doble-ciego, que comparó el tratamiento con lopinavir/ritonavir cápsulas (400/100 mg dos veces al día) más estavudina y lamivudina versus nelfinavir (750 mg TID) más estavudina y lamivudina en 653 pacientes naive a tratamiento antirretroviral. Los pacientes tenían una media de edad de 38 años (rango: 19 a 84), 57% eran caucásicos y el 80% eran hombres. El conteo medio de células CD4 del estadobasal fue de 259 cél/mm3 (rango: 2 a 949 cél/mm3) y la media del RNA VIH-1 plasmático basal fue de 4,9 log10 copias/mL (rango:2,6 a 6,8 log10 copias/mL). La respuesta al tratamiento y el resultado del tratamiento aleatorio se muestran en la Figura 2 y Tabla 11, respectivamente.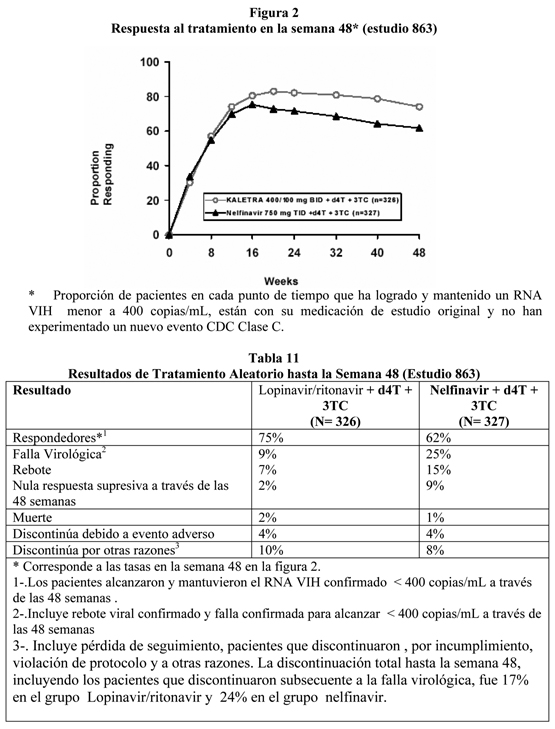
Hasta las 48 semanas de terapia, existió una mayor proporción estadísticamente significativa de pacientes en el grupo lopinavir/ritonavir comparado al grupo nelfinavir con RNA VIHmenor de 400 copias/mL (75% vs. 62%, respectivamente) y RNA VIH menor de 50 copias/mL (67% vs. 52%, respectivamente). La respuesta al tratamiento de los subgrupos en el estado basal de RNA VIH se presenta en la tabla 12.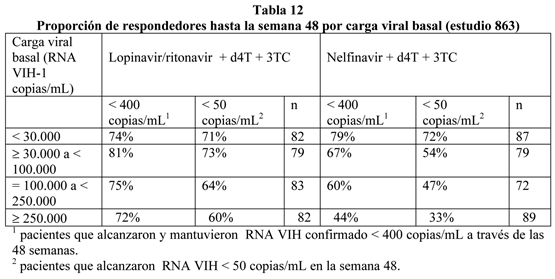
A través de las 48 semanas de terapia, el aumento medio desde el estado basal en el conteo de células CD4 fue de 207 cél/mm3 para el grupo lopinavir/ritonavir y 195 cél/mm3 para el grupo nelfinavir. La figura 3 muestra los estimados de tiempo Kaplan-Meier para falla del tratamiento en el Estudio 863. El tiempo de falla de tratamiento se definió como el tiempo más temprano en que un paciente experimentó falla virológica (dos valores RNA VIH consecutivos demostrando rebote sobre 400 copias/mL), un nuevo evento Clase C CDC, o discontinuación prematura del estudio.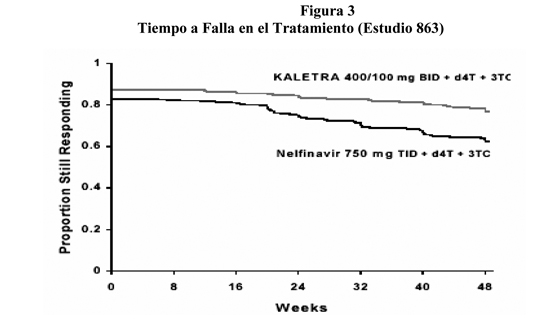
Estudio M97-720: lopinavir/ritonavir cápsulas dos veces al día + estavudina + lamivudina: El estudio M97-720 fue un estudio aleatorio, ciego, multicéntrico que evalúa el tratamiento con lopinavir/ritonavir cápsulas en tres niveles de dosis (Grupo 1: 200/100 mg dos veces al día y 400/100 mg dos veces al día; Grupo II 400/100 mg dos veces al día y 400/200 mg dos veces al día) más lamivudina (150 mg dos veces al día) y estavudina (40 mg dos veces al día) en 100 pacientes. Todos los pacientes se convirtieron a un estudio abierto lopinavir/ritonavir en dosis de 400/100 mg dos veces al día entre las semanas 48 y 72 del estudio. Los pacientes tenían una media de edad de 35 años (rango: 21 a 59), 70% eran caucásicos, y 96% eran hombres. El conteo de células CD4 basal medio fue de 338 cél/mm3 (rango: 3 a 918 céls/mm3) y el RNA VIH-1 plasmático basal medio fue de 4,9 log10 copias/mL (rango: 3,3 a 6,3 log10 copias/mL). A través de las 360 semanas de tratamiento en el estudio 720, la proporción de pacientes con RNA VIH menor de 400 (menor de 50) copias/mL fue 61% (59%) [n=100], y el aumento medio correspondiente en recuento de células CD4 fue 501cél/mm3. Treinta y nueve pacientes (39 %) discontinuaron el estudio, incluyendo 15 (15 %) discontinuaciones debido a eventos adversos y 1 (1%) muerte. Dieciocho pacientes demostraron pérdida de respuesta virológica (dos valores consecutivos de rebote de RNA VIH-1 sobre 400 copias/mL, un valor de rebote de RNA VIH-1 seguido por discontinuación, o falla en alcanzar un RNA VIH < 400 copias/mL). Se realizó análisis genotípico de virus aislados en estos pacientes con valores aislados de RNA VIH-1 > 400 copias/mL después de la semana 24. Habían resultados disponibles de 19 pacientes y confirmaron que no hubo mutaciones en el sitio de acción primario o activo en la proteasa (aminoácidos en las posiciones 8, 30, 32, 36, 47, 48, 50, 82, 84 y 90) o resistencia fenotípica al inhibidor de proteasa. Estudio M02-418: lopinavir/ritonavir cápsulas una vez al día + tenofovir DF + emtricitabina comparado con lopinavir/ritonavir dos veces al día + tenofovir DF + emtricitabina: El estudio 418 fue un estudio randomizado, abierto, multicéntrico comparando el tratamiento con lopinavir/ritonavir cápsulas 800/200 mg una vez al día más tenofovir DF y emtricitabina versus lopinavir/ritonavir cápsulas 400/100 mg dos veces al día más tenofovir DF y emtricitabina en 190 pacientes naive a antirretrovirales. Los pacientes tenían una media de edad de 39 años (rango: 19 a 75), 54% eran caucásicos y 78% eran hombres. El recuento basal medio de células CD4 fue 260 células/mm3 (rango: 3 a 1006 células/mm3) y la media basal de RNA HIV-1 plasmático fue 4.8 log10 copias/mL (gama: 2.6 a 6.4 log10 copias/mL). La respuesta al tratamiento y los resultados del tratamiento randomizado se presentan en la tabla 13.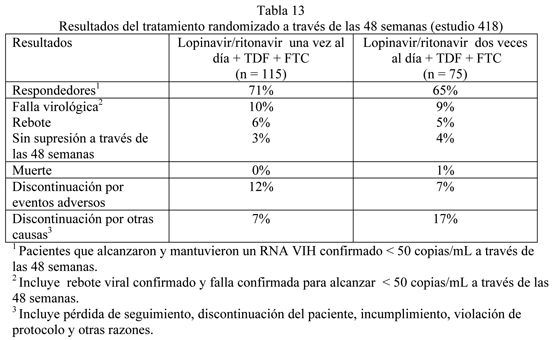
A través de las 48 semanas de terapia, 71% en el grupo lopinavir/ritonavir una vez al día y 65% en el grupo lopinavir/ritonavir dos veces al día alcanzaron y mantuvieron RNA VIH < 50 copias/mL (95% de intervalo de confianza para la diferencia, -7.6% a 19.5%). La media de los aumentos del recuento de células CD4 en la semana 48 fueron 185 células/mm3 para el grupo lopinavir/ritonavir una vez al día y 196 células/mm3 para el grupo lopinavir/ritonavir dos veces al día. Estudio 730: lopinavir/ritonavir comprimidos una vez al día + Tenofovir DF + Emtricitabina comparado con lopinavir/ritonavir comprimidos dos veces al día + Tenofovir DF + Emtricitabina: El estudio 730 fue un estudio randomizado, abierto, multicéntrico que comparaba el tratamiento con lopinavir/ritonavir 800/200 mg una vez al día más tenofovir DF y emtricitabina versus lopinavir/ritonavir 400/100 mg dos veces al día más tenofovir DF y emtricitabina en 664 pacientes naïve a tratamiento antirretroviral. Los pacientes se randomizaron en una razón 1:1 para recibir ya sea lopinavir/ritonavir 800/200 mg una vez (n=333) o lopinavir/ritonavir 400/100 mg dos veces al día (n=331). Una mayor estratificación dentro de cada grupo fue 1:1 (comprimido vs. cápsula). Los pacientes que recibieron la cápsula se cambiaron a la formulación de comprimido en la semana 8 y se mantuvieron en su esquema de dosificación randomizado. A los pacientes se les administró emtricitabina 200 mg una vez al día y tenofovir DF 300 mg una vez al día. La edad media de los pacientes reclutados fue 39 años (rango: 19 a 71); 75% eran Caucásicos, y 78% eran hombres. El recuento de células CD4+ medio en el estado basal fue 216 células/mm3 (rango: 20 a 775 células/mm3) y la media del RNA VIH-1 plasmático en el estado basal fue 5.0 log10 copias/mL (rango: 1.7 a 7.0 log10 copias/mL). La respuesta al tratamiento y los resultados del tratamiento randomizado hasta la semana 48 se presentan en la Tabla 14.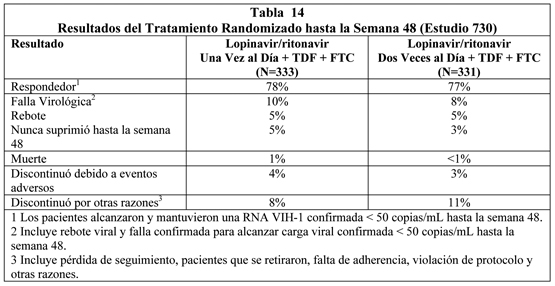
Hasta las 48 semanas de terapia, 78% en el brazo lopinavir/ritonavir una vez al día y 77% en el brazo lopinavir/ritonavir dos veces al día alzanzaron y mantuvieron RNA VIH-1 < 50 copias/mL (95% intervalo de confianza para la diferencia, -5.9% a 6.8%). La media del aumento del recuento de células CD4+ en la semana 48 fue 186 células/mm3 para el brazo lopinavir/ritonavir una vez al día y 198 células/mm3 para el brazo lopinavir/ritonavir dos veces al día. Pacientes con Terapia Antiretroviral Previa: Estudio M98-888: lopinavir/ritonavir cápsulas dos veces al día + nevirapina + NRTIs comparado con inhibidores de proteasa seleccionados por el investigador + nevirapina + NRTIs: El estudio 888 fue un estudio randomizado, abierto, multicéntrico que comparaba el tratamiento con lopinavir/ritonavir cápsulas (400/100 mg dos veces al día) más nevirapina y nucleósidos inhibidores de transcriptasa reversa versus inhibidores de proteasa seleccionados por el investigador más nevirapina y nucleósidos inhibidores de transcriptasa reversa en 288 pacientes que han utilizado un inhibidor de proteasa, naive a inhibidor no nucleósido de transcriptasa reversa (NNRTI). Los pacientes tenían una media de edad de 40 años (rango: 18 a 74), 68% eran caucásicos, y 86% eran hombres. El recuento basal medio de células CD4 fue 322 celulas/mm3 (rango: 10 a 1059 células/mm3) y la media basal de RNA VIH-1 plasmático fue 4.1 log10 copias/mL (rango: 2.6 a 6.0 log10 copias/mL). La respuesta al tratamiento y los resultados del tratamiento randomizado a través de las 48 semanas se presentan en la Figura 4 y la Tabla 15, respectivamente.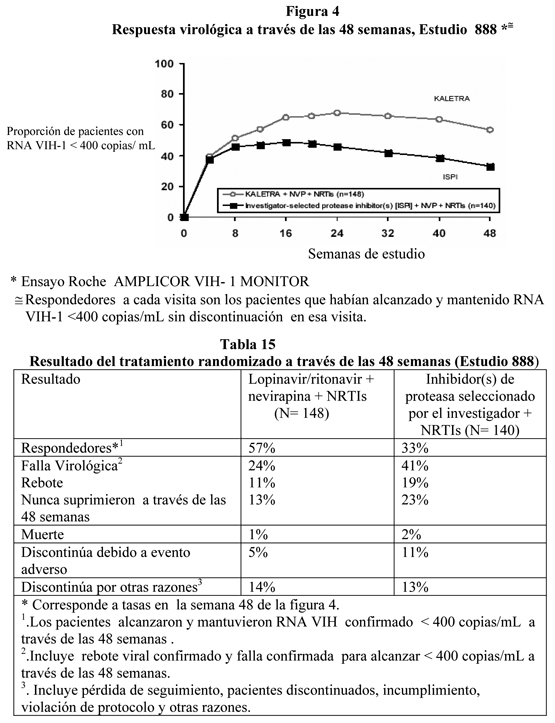
Estudio M97-765: lopinavir/ritonavir cápsulas dos veces al día + nevirapina + NRTIs: El estudio M97-765 era un estudio aleatorio, ciego, multicéntrico que evaluó el tratamiento con lopinavir/ritonavir cápsulas en dos niveles de dosis (400/100 mg dos veces al día y 400/200 mg dos veces al día) más nevirapina (200 mg dos veces al día) y dos NRTIs en 70 pacientes que han utilizado un inhibidor de proteasa, naive a inhibidor no nucleósido de transcriptasa reversa (NNRTI). Los pacientes tenían una media de edad de 40 años (rango 22 a 66 años), 73% eran caucásicos, y 90% hombres. El conteo medio de células CD4 basal fue 372 cél/mm3 (rango 72 a 807 cél/mm3) y RNA VIH-1 plasmático basal medio fue 4.0 log10 copias/mL (rango 2,9 a 5,8 log10 copias/mL). A través de las 144 semanas de tratamiento en el estudio 765, la proporción de pacientes con RNA VIH menor de 400 (menor de 50) copias/mL fue 54% (50%) [n=70], y el aumento medio correspondiente del recuento de células CD4 fue 212 cél/mm3. Veintisiete pacientes (39%) discontinuaron el estudio, incluyendo 9 discontinuaciones (13%) secundarias a eventos adversos y 2 (3%) a muertes. Estudio 802: Lopinavir/ritonavir comprimidos 800/200 Una Vez al Día Versus 400/100 mg Dos Veces al Día cuando se Coadministra con Inhibidores Nucleósido/Nucleótido de Transcriptasa Reversa en Sujetos Infectados VIH-1 con Experiencia con Antiretroviral: El estudio M06-802 fue un estudio randomizado, abierto, multicéntrico que comparaba la seguridad, tolerabilidad y actividad antiviral en una dosificación una vez al día y dos veces al día de lopinavir/ritonavir comprimidos en 599 sujetos con cargas virales detectables mientras recibieron su terapia antirretroviral actual. Los pacientes se randomizaron en una razón 1:1 para recibir ya sea lopinavir/ritonavir 800/200 mg una vez al día (n=300) o lopinavir/ritonavir 400/100 mg dos veces al día (n=299). Los pacientes recibieron al menos dos inhibidores nucleósido/nucleótido de transcriptasa reversa seleccionado por el investigador. La media de edad de los pacientes reclutados fue 41 años (rango: 21 a 73); 51% eran Caucásicos y 66% eran hombres. El recuento de células CD4+ medio en el estado basal fue 254 células/mm3 (rango: 4 a 952 células/mm3) y la media del RNA VIH-1 plasmático en el estado basal fue 4.3 log10 copias/mL (rango: 1.7 a 6.6 log10 copias/mL). La respuesta al tratamiento y los resultados del tratamiento randomizado hasta la semana 48 se presentan en la Tabla 16.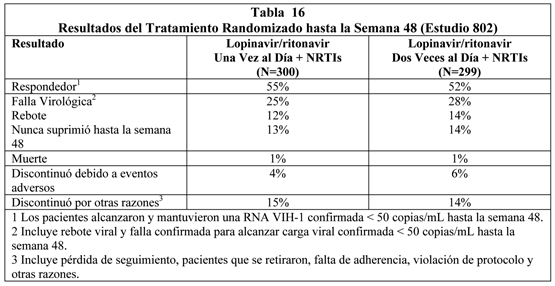
Uso Pediátrico: Estudio M98-940: El estudio M98-940 fue un estudio abierto, multicéntrico que evalúa el perfil farmacocinético, tolerabilidad, seguridad y eficacia de la solución oral de lopinavir/ritonavir que contiene lopinavir 80 mg/mL y ritonavir 20 mg/mL en 100 pacientes pediátricos naive a antirretrovirales (44%) y que los han utilizado (56%). Todos los pacientes eran naive a inhibidor no-nucleósido de transcriptasa reversa. Los pacientes eran escogidos al azar para dosis de 230 mg lopinavir/57,5 mg ritonavir por m2 o 300 mg de lopinavir/75 mg de ritonavir por m2. Los pacientes naive también recibieron lamivudina y estavudina. Los pacientes que ya lo habían utilizado recibieron nevirapina más hasta dos nucleósidos inhibidores de transcriptasa reversa. La seguridad, eficacia y perfiles farmacocinéticos de los dos regímenes de dosis se evaluaron después de tres semanas de terapia en cada paciente. Después del análisis de estos datos, todos los pacientes continuaron con dosis de 300 mg de lopinavir/75mg ritonavir m2. Los pacientes tenían una media de edad de cinco años (rango seis meses a 12 años) con un 14% menores de dos años. El conteo de células CD4 medio basal fue 838 cél/mm3 y un RNA VIH-1 plasmático basal medio de 4.7 log10 copias/mL. A través de las 48 semanas de terapia, la proporción de pacientes que alcanzaron y sostuvieron un H RNA VIH menos de 400 copias/mL fue 80% para pacientes naive a antirretrovirales y 71% para pacientes que han utilizado antirretrovirales. El aumento del conteo de células CD4 medio basal fue 404 cél/mm3 para naive a antirretrovirales y 284 cél/mm3 para pacientes que han utilizado antirretrovirales a través de las 48 semanas. Se observó discontinuaciones prematuras en 2 (2%) sujetos previo a las 48 semanas. Uno de éstos fue considerado por el investigador como "no relacionado" con la droga del estudio, el segundo "posiblemente" relacionado con la droga del estudio. La selección de la dosis para pacientes entre 6 meses a 12 años se basa en los siguientes resultados. El régimen 230/57.5 mg/m2 dos veces al día sin nevirapina y el régimen 300/75 mg/m2 dos veces al día con nevirapina proporcionó concentraciones plasmáticas de lopinavir similares a las obtenidas en pacientes adultos que recibieron el régimen 400/100 mg dos veces al día (sin nevirapina).
Indicaciones.
Lopinavir/ritonavir está indicado en combinación con otros agentes antirretrovirales para el tratamiento de la infección por VIH. Esta indicación está basada en análisis de niveles de ARN VIH plasmático y conteo de células CD4 en un estudio controlado de lopinavir/ritonavir de 48 semanas de duración y en estudios más pequeños de rango de dosis no controlados de lopinavir/ritonavir de 144 semanas de duración. En la actualidad, no existen resultados de los estudios controlados que evalúan el efecto de lopinavir/ritonavir en la progresión clínica del VIH.
Dosificación.
Adultos: Los
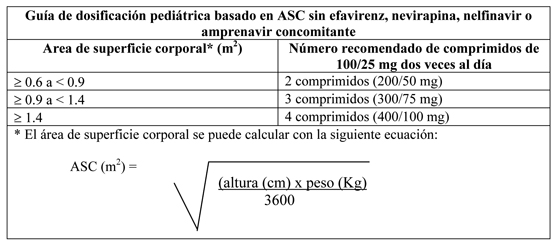
Terapia concomitante: Efavirenz, nevirapina, nelfinavir o (fos)amprenavir: La siguiente tabla contiene la guía de dosificación para lopinavir/ritonavir comprimidos 100/25 mg basado en ASC cuando se usa en combinación con efavirenz, nevirapina, nelfinavir o (fos)amprenavir en niños:
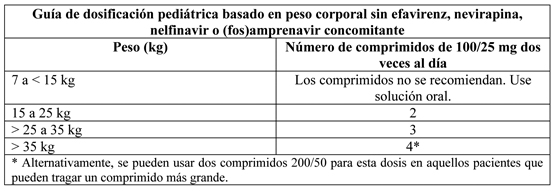
La siguiente tabla contiene la guía de dosificación para lopinavir/ritonavir comprimidos 100/25 mg basado en peso corporal:
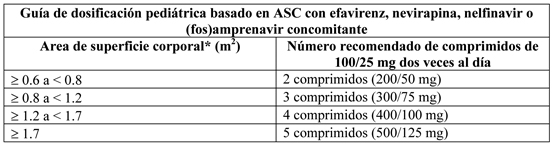
Terapia concomitante: Efavirenz, nevirapina, nelfinavir o (fos)amprenavir: La siguiente tabla contiene la guía de dosificación para lopinavir/ritonavir comprimidos 100/25 mg basado en peso corporal cuando se usa en combinación con efavirenz, nevirapina, nelfinavir o amprenavir en niños:
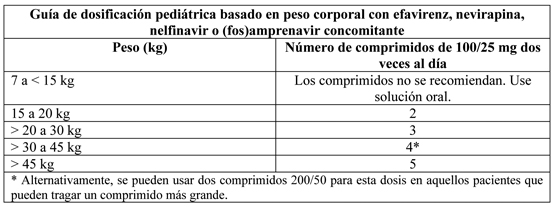
Kaletra solución oral está disponible para los pacientes que no pueden tomar una formulación de comprimidos. Por favor ver Folleto de Información al Profesional de Kaletra Solución Oral para instrucciones de dosificación.
Contraindicaciones.
Lopinavir/ritonavir está contraindicado en pacientes con hipersensibilidad conocida a lopinavir, ritonavir o cualquiera de sus excipientes. lopinavir/ritonavir no se debe co-administrar concurrentemente con drogas que son altamente dependientes de CYP3A para su clearance y para las concentraciones plasmáticas elevadas que se asocian con eventos serios y/o potencialmente mortales. Estas drogas se presentan en la Tabla 1.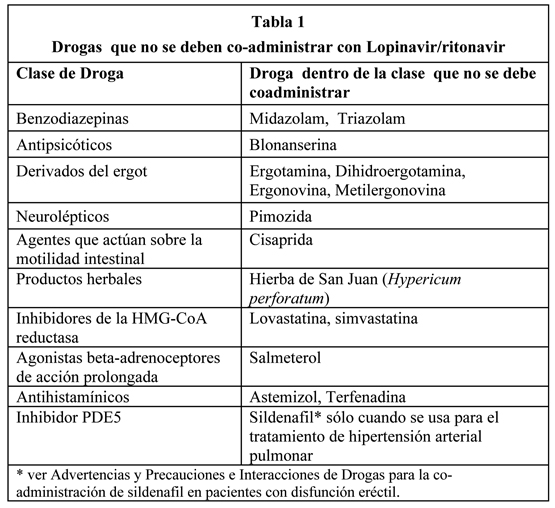
Embarazo y lactancia.
Embarazo, Fertilidad y Reproducción: Lopinavir en combinación con ritonavir en una proporción 2 a 1 no produjo ningún efecto sobre fertilidad en las ratas machos y hembras en las dosis máximas alcanzables produciendo exposiciones a la droga las cuales eran comparables a o levemente menores que aquellas alcanzadas con la dosis terapéutica recomendada, niveles de 10/5, 30/15 o 100/50 mg/kg/día. Basado en las medidas de AUC, las exposiciones en ratas en dosis altas eran aproximadamente 0.7-veces para lopinavir y 1.8-veces para ritonavir de las exposiciones en seres humanos en la dosis terapéutica recomendada (400/100mg dos veces al día). No se observaron malformaciones relacionadas con el tratamiento cuando lopinavir/ritonavir fue administrado a ratas o conejas preñadas. La toxicidad de desarrollo embrionario y fetal observada en ratas (resorción temprana, viabilidad fetal disminuida, peso corporal fetal disminuido, aumento de la incidencia de variaciones esqueléticas y retraso en la osificación esquelética) ocurrió en ratas en una dosificación maternalmente tóxica (100/50 mg/kg/día). Basado en las medidas de AUC, las exposiciones de la droga en ratas de 100/50 mg/kg/día eran aproximadamente 0.7 veces para lopinavir y 1.8 veces para ritonavir para los machos y hembras que de las exposiciones en seres humanos en la dosis terapéutica recomendada (400/100mg dos veces al día). En un estudio peri- y postnatal en ratas, una toxicidad de desarrollo (una disminución en la sobrevida de las crías entre el nacimiento y el día 21 postnatal) ocurrió con 40/20 mg/kg/día y más. No se observó ninguna toxicidad de desarrollo embrionario y fetal en conejos en una dosificación maternalmente tóxica (80/40 mg/kg/día). Basado en las medidas del AUC, las exposiciones de la droga en conejos en 80/40 mg/kg/día fueron aproximadamente 0.6-veces para lopinavir y 1.0 veces para ritonavir que las exposiciones en seres humanos en la dosis terapéutica recomendada (400/100mg dos veces al día). No hay, sin embargo, estudios adecuados y bien controlados en mujeres embarazadas. Debido a que los estudios de reproducción en animales no siempre predicen la respuesta en los seres humanos, lopinavir/ritonavir se debe usar durante el embarazo sólo si los beneficios potenciales justifican los riesgos potenciales para el feto. Uso durante la lactancia: Debido tanto al potencial de transmisión del VIH como al potencial de reacciones adversas serias en los lactantes, se debe aconsejar a las madres no amamantar a sus hijos mientras estén recibiendo lopinavir/ritonavir. Estudios en ratas han demostrado que el lopinavir es secretado en la leche. No se sabe si el lopinavir se secreta en la leche materna.
Reacciones adversas.
Adultos: Eventos Adversos Tratamientos Emergentes: lopinavir/ritonavir se ha estudiado en 2.154 pacientes infectados con VIH-1 como terapia combinada en estudios clínicos Fase I/II y Fase III. El evento adverso más común asociado con la terapia de lopinavir/ritonavir fue diarrea, la cual generalmente fue de leve a moderada severidad. En el Estudio 863, las tasas de discontinuación en terapia aleatoria debido a eventos adversos, que incluyeron muerte, fueron 5,8% en pacientes tratados con lopinavir/ritonavir y 4,9% con nelfinavir. La incidencia de diarrea fue mayor para lopinavir/ritonavir cápsulas una vez al día comparado con lopinavir/ritonavir cápsulas dos veces al día en el estudio 418 (ver Tabla 2). Los eventos adversos tratamiento-emergentes de intensidad moderada a severa en mayor que o igual a 2% de los pacientes tratados con terapia combinada incluyendo lopinavir/ritonavir por hasta 48 semanas (estudios 863, 418 y 730) y por hasta 360 semanas (estudio 720) se presentan en la Tabla 2 (pacientes naïve a antirretrovirales) y por hasta 48 semanas (estudios 888 y 802), 8 4 semanas (estudio 957) y 144 semanas (estudio 765) en la Tabla 3 (pacientes con experiencia con antirretrovirales). Para otra información con respecto a eventos adversos observados o potencialmente serios, ver por favor Advertencias.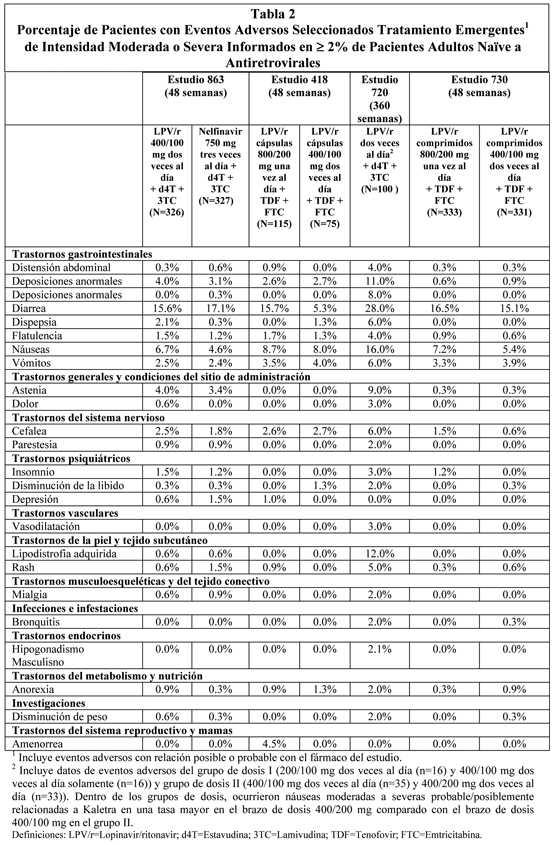
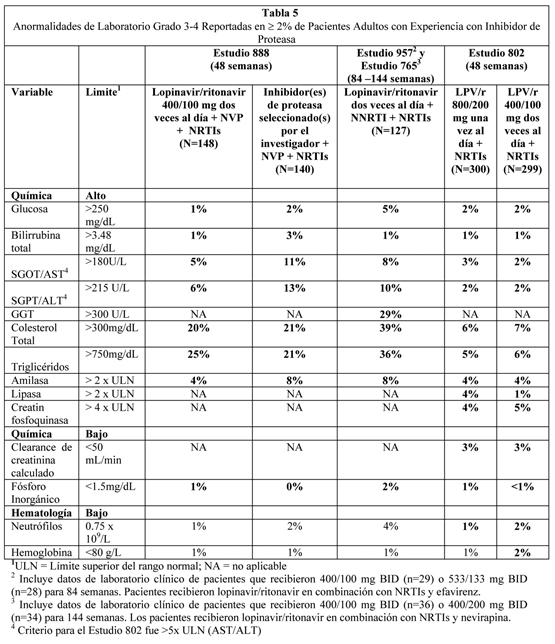
Los eventos adversos tratamiento emergentes que ocurrieron en menos del 2% de los pacientes adultos tratados que recibieron lopinavir/ritonavir en todos los estudios clínicos de Fase II/III, y considerados al menos posiblemente relacionados o de relación desconocida con el tratamiento con lopinavir/ritonavir y al menos de moderada intensidad, se describen abajo por sistema órgano clase. Infecciones e infestaciones: Infección bacteriana, bronconeumonia, celulitis, foliculitis, furunculosis, gastroenteritis, influenza, otitis media, absceso perineal, faringitis, rinitis, sialoadenitis, sinusitis, e infección viral. Neoplasias benigna, maligna e inespecífica (incluyendo quistes y pólipos): Neoplasia y neoplasia benigna de la piel y neoplasia. Trastornos del sistema sanguíneo y linfático: Anemia, leucopenia, y linfadenopatía, neutropenia y esplenomegalia. Trastornos del sistema inmune: Hipersensibilidad a drogas, hipersensibilidad y síndrome de reconstitución inmune. Trastornos endócrinos: Síndrome de Cushing e hipotiroidismo. Trastornos del metabolismo y nutrición: Disminución del apetito, deshidratación, diabetes mellitus, hiperamilasemia, hiperlipasemia, hipovitaminosis, aumento del apetito, acidosis láctica, lipomatosis y obesidad. Trastornos psiquiátricos: Sueños anormales, labilidad afectiva, agitación, ansiedad, apatía, estado confusional, desorientación, cambios del ánimo, nerviosismo y pensamiento anormal. Trastornos del sistema nervioso: Ageusia, amnesia, trastornos del equilibrio, ataxia, infarto cerebral, convulsión, vértigo, disgeusia, diskinesia, encefalopatía, trastorno extrapiramidal, parálisis facial, hipertonía, migraña, neuropatía, neuropatía periférica, somnolencia y temblor. Trastornos de la visión: Trastorno del ojo y alteración visual. Trastornos del oído y del laberinto: Hiperacusia, tinnitus y vértigo. Trastornos cardíacos: Angina pectoris, fibrilación auricular, bloqueo atrioventricular, infarto al miocardio, palpitación e incompetencia de la válvula tricúspide. Trastornos vasculares: Trombosis venosa profunda, hipotensión ortostática, tromboflebitis, várices y vasculitis. Trastornos respiratorios, torácico y del mediastino: Asma, tos, disnea y edema pulmonar. Trastornos gastrointestinales: Disconfort abdominal, dolor de abdomen inferior, constipación, boca seca, duodenitis, enteritis, enterocolitis, enterocolitis hemorrágica, eructos, esofagitis, incontinencia fecal, gastritis, úlcera gástrica, enfermedad de reflujo gastroesofágico, hemorroides, ulceración de la boca, pancreatitis, periodontitis, hemorragia rectal y estomatitis. Trastornos Hepatobiliares: Colangitis, colecistitis, esteatosis hepática, hepatitis, hepatomegalia, ictericia, depósitos grasos en el hígado y sensibilidad hepática. Trastornos de la piel y tejido subcutáneo: Acné, alopecia, dermatitis alérgica, dermatitis exfoliativa, piel seca, eczema, hiperhidrosis, capilaritis idiopática, trastornos de las uñas, prurito, rash maculopapular, seborrea, decoloración de la piel, hipertrofia de la piel, piel estriada, úlcera de piel e inflamación de la cara. Trastornos Musculoesqueléticos y del tejido conectivo: Artralgia, artropatía, artrosis, dolor de espalda, debilidad muscular, osteoartritis, osteonecrosis y dolor en la extremidad. Trastornos renales y urinarios: Hematuria, nefritis, nefrolitiasis, anormalidad de la orina y olor anormal de la orina. Trastornos del sistema reproductivo y mamas: Agrandamiento de mamas, trastorno de la eyaculación, disfunción eréctil, ginecomastia y menorragia. Trastornos generales y condiciones del sitio de administración: Dolor de pecho, quiste, interacción de droga, edema, edema periférico, edema facial, hipertrofia y malestar. Investigaciones: Aumento del nivel de la droga y disminución de la tolerancia a la glucosa y aumento de peso. Anormalidades de Laboratorio: La Tabla 4 y 5 presentan el porcentaje de pacientes adultos tratados con la terapia de combinación que incluyó lopinavir/ritonavir con anormalidades de laboratorio Grado 3 a 4.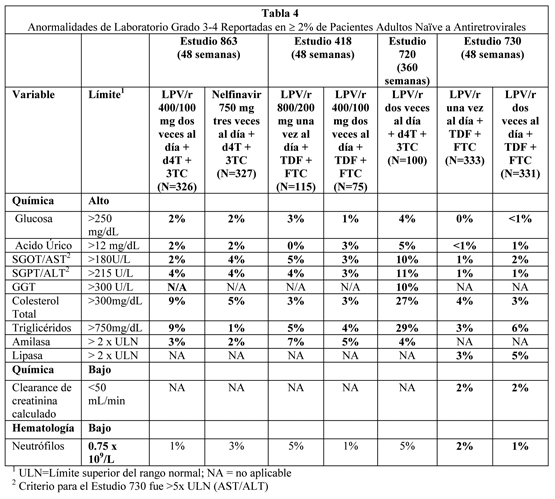
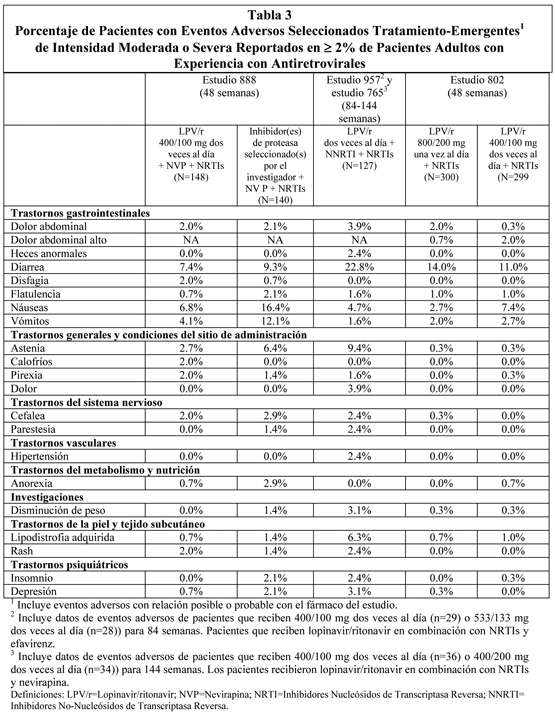
Pediatría: Eventos Adversos Tratamiento Emergentes: lopinavir/ritonavir se ha estudiado en 100 pacientes pediátricos entre los 6 meses y los 12 años de edad. El perfil de eventos adversos observados durante un estudio clínico fue similar al de los pacientes adultos. Disgeusia, vómitos y diarrea fueron los eventos adversos más comúnmente reportados de cualquier severidad en relación a la droga en pacientes pediátricos tratados con terapia combinada incluyendo lopinavir/ritonavir por hasta 48 semanas en el estudio 940. Un total de 8 niños experimentaron eventos adversos moderados a severos al menos posiblemente relacionados con lopinavir/ritonavir. Rash (registrado en un 3%) fue el único evento adverso de intensidad moderada a severa relacionado con la droga en ≥2% de los niños reclutados. Anormalidades de Laboratorio: La Tabla 6 presenta los porcentajes de pacientes pediátricos tratados con la terapia combinada con lopinavir/ritonavir con anormalidades de laboratorio Grado 3 a 4.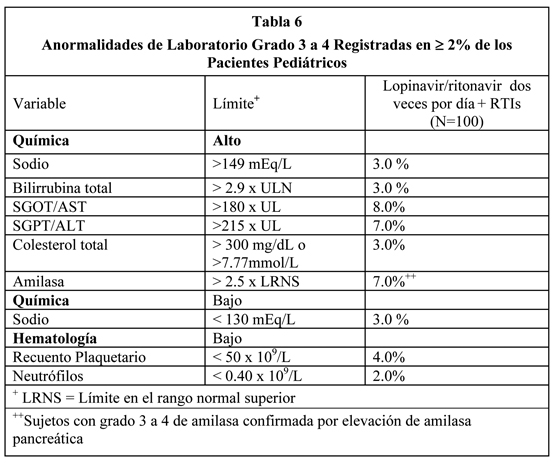
Experiencia Post-marketing: Se ha reportado hepatitis en pacientes en tratamiento con lopinavir/ritonavir. Se ha reportado necrólisis tóxica epidérmica, síndrome de Stevens-Johnson y eritema multiforme. Se ha reportado bradiarritmia.
Advertencias.
Interacciones de Droga: lopinavir/ritonavir es un inhibidor de la isoforma CYP3A del citocromo P450. La co-administración de lopinavir/ritonavir con otras drogas principalmente metabolizadas por CYP3A puede provocar una elevación de las concentraciones plasmáticas de la otra droga que podría aumentar o prolongar sus efectos terapéuticos y adversos (ver Contraindicaciones -Tabla 1, Interacciones de droga, y Farmacocinética: Interacciones Droga-Droga). Antimicobacterianos: lopinavir/ritonavir en dosis estándar no se debe co-administrar con rifampicina debido a que grandes disminuciones en las concentraciones de lopinavir pueden significativamente disminuir el efecto terapéutico (ver Interacciones de droga). Corticoesteroides: El uso concomitante de lopinavir/ritonavir y de propionato de fluticasona puede aumentar significativamente las concentraciones plasmáticas del propionato de fluticasona y reducir las concentraciones séricas de cortisol. Se han reportado efectos sistémicos del corticoesteroide que incluyen el síndrome de Cushing y la supresión suprarrenal cuando el ritonavir se ha co-administrado con propionato de fluticasona inhalado o intranasal. Hallazgos similares con la administración concomitante de Lopinavir/ritonavir y otros corticoesteroides inhalados que se metabolizan similarmente a la fluticasona, por ejemplo budesonida, no se pueden excluir. Particular precaución se debe tener al utilizar Lopinavir/ritonavir y cualquiera de estos glucocorticoides administrados por inhaladores o intranasalmente (ver Interacciones de droga). Agentes para la disfunción eréctil (inhibidores PDE5): Particular precaución se debe tener al prescribir sildenafil, tadalafil o vardenafil para el tratamiento de la disfunción eréctil en pacientes en tratamiento con lopinavir/ritonavir. La co-administración de lopinavir/ritonavir con estas drogas puede aumentar sustancialmente las concentraciones de éstas y puede provocar un aumento en los eventos adversos asociados por ejemplo, hipotensión, y erección prolongada. El uso concomitante de sildenafil con lopinavir/ritonavir está contraindicado en pacientes con hipertensión arterial pulmonar (ver Contraindicaciones e Interacciones de droga). Productos herbales: Pacientes en tratamiento con lopinavir/ritonavir no deben usar productos que contengan Hierba de San Juan (Hypericum perforatum) porque la co-administración puede reducir las concentraciones plasmáticas de los inhibidores de proteasa. Esto puede ocasionar pérdida del efecto terapéutico y desarrollo de resistencia a lopinavir o a la clase terapéutica de los inhibidores de proteasa (ver Contraindicaciones e Interacciones de droga). Inhibidores de HMG-CoA reductasa: El uso concomitante de lopinavir/ritonavir con lovastatina o simvastatina está contraindicado (ver Contraindicaciones). Se debe tener precaución si inhibidores de proteasa VIH, incluyendo lopinavir/ritonavir, se usan concurrentemente con rosuvastatina o con otros inhibidores de HMG-CoA reductasa que también son metabolizados por la vía CYP3A4 (ej., atorvastatina), ya que esto puede aumentar el potencial de reacciones serias tales como, miopatía, incluyendo rabdomiólisis (ver Interacciones de droga). Tipranavir: En un estudio clínico de terapia de combincación de inhibidor de proteasa con booster dual en adultos VIH positivos que han utilizado múltiples tratamientos, tipranavir (500 mg dos veces al día) con ritonavir (200 mg dos veces al día), co-administrados con lopinavir/ritonavir (400/100 mg dos veces al día) produjeron 47% y 70% de reducción del AUC y Cmín de lopinavir, respectivamente. Por lo tanto, la administración concomitante de lopinavir/ritonavir y tipranavir con baja dosis de ritonavir no se recomienda. Diabetes Mellitus/Hiperglucemia: Se han informado diabetes mellitus de nuevo inicio, exacerbación de diabetes mellitus preexistente e hiperglucemia durante la vigilancia post-comercialización en pacientes infectados con VIH que reciben terapia con inhibidor de proteasa. Algunos pacientes requirieron inicio o ajuste de la dosis de insulina o agentes hipoglucemiantes orales para el tratamiento de estos eventos. En algunos casos, ha ocurrido cetoacidosis diabética. En aquellos pacientes que discontinuaron la terapia con inhibidor de proteasa, la hiperglucemia persistió en algunos casos. Debido a que estos eventos han sido informados voluntariamente durante la práctica clínica, no se pudo estimar su frecuencia y no se ha establecido la relación causal entre la terapia con inhibidor de proteasa y estos eventos. Pancreatitis: Se ha observado pancreatitis en pacientes tratados con lopinavir/ritonavir, incluyendo a aquellos que desarrollaron marcadas elevaciones de triglicéridos. Se han observado decesos en algunos casos. Aunque no se ha establecido una relación causal con lopinavir/ritonavir, las marcadas elevaciones de triglicéridos son un factor de riesgo para el desarrollo de pancreatitis (ver Advertencias: Elevaciones de Lípidos). Los pacientes con enfermedad por VIH avanzada pueden tener un riesgo aumentado de triglicéridos elevados y pancreatitis, y pacientes con una historia de pancreatitis pueden tener un aumento del riesgo de recurrencia durante el tratamiento con lopinavir/ritonavir. Insuficiencia Hepática: lopinavir/ritonavir se metaboliza principalmente en el hígado. Por lo tanto, se debe tener precaución cuando se administra esta droga a pacientes con función hepática alterada. Los datos farmacocinéticos sugieren aumentos en concentraciones plasmáticas de lopinavir de aproximadamente 30%, así como disminuciones de la unión a proteínas plasmáticas en pacientes con VIH y co-infectados con HVC con alteración hepática leve a moderada (ver Farmacología: Farmacocinética). Los pacientes con hepatitis B o C subyacente o elevaciones marcadas en las transaminasas antes del tratamiento pueden estar en mayor riesgo de desarrollar elevaciones futuras de las transaminasas. Ha habido reportes postmarketing de disfunción hepática, incluyendo algunas muertes. Estos han ocurrido generalmente en pacientes con enfermedad VIH avanzada que tomaban múltiples medicamentos concomitantes en un escenario de hepatitis crónica o cirrosis subyacente. Una relación causal con la terapia de lopinavir/ritonavir no se ha establecido. Un aumento de la supervisión de AST/ALT se debe considerar en estos pacientes, especialmente durante los primeros meses de tratamiento con lopinavir/ritonavir. Resistencia/Resistencia Cruzada: Se han observado distintos grados de resistencia cruzada entre los inhibidores de proteasa. Se está investigando el efecto del tratamiento con lopinavir/ritonavir sobre la eficacia de inhibidores de proteasa administrados posteriormente (ver Farmacología: Microbiología). Hemofilia: Se ha informado aumento de sangrado, incluyendo hematomas espontáneos de la piel y hemartrosis, en pacientes con hemofilia tipo A y B tratados con inhibidores de proteasa. En algunos pacientes, se administró factor VIII adicional. En más de la mitad de los casos informados, se continuó o reanudó el tratamiento con inhibidores de proteasa. No se ha establecido una relación causal o un mecanismo de acción entre el tratamiento con inhibidor de proteasa y estos eventos. Prolongación del Intervalo PR: Se ha mostrado que lopinavir/ritonavir causa una modesta prolongación asintomática del intervalo PR en algunos pacientes. Raros informes de bloqueo atrioventricular de segundo o tercer grado se han informado en pacientes con enfermedad cardíaca estructural subyacente y anormalidades pre-existentes del sistema de conducción o en pacientes que reciben drogas conocidas que prolongan el intervalo PR (tales como verapamilo o atazanavir), que reciben lopinavir/ritonavir. Lopinavir/ritonavir se debe usar con precaución en tales pacientes (ver Farmacología). Redistribución de la grasa: En pacientes tratados con antirretrovirales se ha observado una redistribución/acumulación de la grasa corporal que incluye obesidad central, aumento de la grasa dorso-cervical (joroba de búfalo), consunción periférica, consunción facial, hipertrofia mamaria y "aspecto cushingoide". Se desconoce hasta el momento el mecanismo y las consecuencias a largo plazo de estos eventos. No se ha establecido una relación causal. Elevaciones de lípidos: El tratamiento con lopinavir/ritonavir ha producido aumento en la concentración de colesterol total y triglicéridos (ver Reacciones adversas - Tablas 4 y 5). Se deben realizar análisis de triglicéridos y colesterol antes de iniciar el tratamiento con lopinavir/ritonavir y determinaciones periódicas durante el mismo. Los trastornos lipídicos se deben manejar como sea apropiado clínicamente. Ver Advertencias, Inhibidores de HMG-CoA reductasa para información adicional sobre interacciones potenciales de la droga con lopinavir/ritonavir e inhibidores de la HMG CoA reductasa. Síndrome de reconstitución inmune: Se ha reportado el síndrome de reconstitución inmune en pacientes infectados con HIV tratados con combinación de terapia antirretroviral, incluyendo lopinavir/ritonavir. Durante la fase inicial del tratamiento combinado antirretroviral, cuando el sistema inmune responde, los pacientes pueden desarrollar una respuesta inflamatoria a infecciones oportunistas asintomáticas o residuales (tales como infección por Mycobacterium avium, citomegalovirus, neumonía por Pneumocystis jiroveci, o tuberculosis), que pueden hacer necesario evaluación y tratamiento adicionales. Uso en Geriatría: Los estudios clínicos con lopinavir/ritonavir no incluyeron un número suficiente de sujetos de 65 años y más para determinar si responden en forma diferente a los sujetos más jóvenes. En general, se recomienda precaución en la administración de lopinavir/ritonavir y monitoreo de pacientes geriátricos por su mayor frecuencia de disfunción hepática, renal o cardíaca y de enfermedades concomitantes u otra terapia con drogas. Uso en pediatría: No se han establecido los perfiles de seguridad y farmacocinético de lopinavir/ritonavir en pacientes pediátricos de menos de 6 meses de vida. En pacientes VIH infectados de 6 meses a 12 años de edad, el perfil de eventos adversos observado durante un estudio clínico fue similar al de los pacientes adultos. Se encuentran en curso estudios clínicos que evalúan la actividad antiviral de lopinavir/ritonavir en pacientes pediátricos (ver Descripción de estudios clínicos). lopinavir/ritonavir una vez al día no se ha evaluado en pacientes pediátricos.
Interacciones.
Lopinavir/ritonavir es un inhibidor de CYP3A tanto in vitro como in vivo. La co-administración de lopinavir/ritonavir y otras drogas principalmente metabolizadas por CYP3A (por ejemplo, bloqueadores de los canales de calcio dihidropiridínicos, inhibidores de la HMG-CoA reductasa, inmunosupresores e inhibidores PDE5) puede elevar las concentraciones plasmáticas de otras drogas que podrían aumentar o prolongar sus efectos terapéuticos y adversos (ver Advertencias e Interacciones de droga). Los agentes que son extensamente metabolizados por CYP3A y que tienen un alto metabolismo de primer paso parecen ser los más susceptibles a sufrir un gran aumento en su AUC ( > 3 veces) cuando se co-administran con lopinavir/ritonavir. Las drogas que están específicamente contraindicadas debido a la magnitud esperada de interacción y al potencial de eventos adversos serios, se listan en la Tabla 1 en Contraindicaciones. lopinavir/ritonavir es metabolizado por la CYP3A. La co-administración de lopinavir/ritonavir y drogas que inducen CYP3A puede disminuir las concentraciones plasmáticas de lopinavir y reducir su efecto terapéutico (ver Advertencias e Interacciones de droga). Si bien no se ha observado este efecto con ketoconazol concurrente, la co-administración de lopinavir/ritonavir y otras drogas que inhiben CYP3A puede aumentar las concentraciones plasmáticas de lopinavir. Agentes anti-VIH: Inhibidores Nucléosidos de Transcriptasa Reversa (NRTIs): Estavudina y Lamivudina: No se observaron variaciones en la farmacocinética de lopinavir cuando se administró lopinavir/ritonavir solo o en combinación con estavudina y lamivudina. Didanosina: Se recomienda que la didanosina se administre con el estómago vacío; por lo tanto, la didanosina se puede co-administrar con los comprimidos de lopinavir/ritonavir sin alimentos. Zidovudina y Abacavir: lopinavir/ritonavir induce glucuronización, por lo tanto tiene el potencial para reducir las concentraciones plasmáticas de zidovudina y abacavir. Se desconoce la significancia clínica de esta interacción potencial. Tenofovir: Un estudio ha mostrado que lopinavir/ritonavir aumenta las concentraciones de tenofovir. El mecanismo de esta interacción es desconocido. Los pacientes que reciben lopinavir/ritonavir y tenofovir deben ser supervisados por los eventos adversos asociados a tenofovir. Todos: Se ha reportado incremento de la CPK (creatin fosfoquinasa), mialgia, miositis y raramente rabdomiólisis con los inhibidores de proteasa, particularmente en combinación con NRTIs (inhibidores nucleósidos de transcriptasa reversa). Inhibidores No Nucleósidos de Transcriptasa Reversa (NNRTIs): Nevirapina: No hubo cambios aparentes en la farmacocinética de lopinavir en sujetos adultos sanos durante la co-administración de nevirapina y lopinavir/ritonavir. Los resultados de un estudio en sujetos pediátricos VIH-positivos revelaron una disminución de las concentraciones de lopinavir durante la co-administración de nevirapina. Es de esperar que el efecto de la nevirapina en adultos VIH-positivos sea similar al observado en sujetos pediátricos y que por lo tanto pueden disminuir las concentraciones de lopinavir. Se desconoce la significancia clínica de esta interacción farmacocinética. lopinavir/ritonavir no se debe administrar una vez al día en combinación con nevirapina. Efavirenz: El aumento de la dosis de lopinavir/ritonavir a 500/125 mg 2 veces por día produjo concentraciones plasmáticas de lopinavir similares comparado con lopinavir/ritonavir comprimidos 400/100 mg dos veces al día sin efavirenz (ver Dosificación y Tabla 9). El aumento de la dosis de lopinavir/ritonavir a 600/150 mg (tres (3) comprimidos) 2 veces por día co-administrado con efavirenz aumentó significativamente las concentraciones plasmáticas de Lopinavir y Ritonavir en aproximadamente un 36% y 56% a 92%, respectivamente comparado con lopinavir/ritonavir comprimidos 400/100 mg dos veces al día sin efavirenz (ver Tabla 9). Nota: El efavirenz y la nevirapina inducen actividad de CYP3A y de este modo tienen el potencial de disminuir las concentraciones plasmáticas de otros inhibidores de proteasa cuando se usan en combinación con lopinavir/ritonavir. lopinavir/ritonavir no se debe administrar una vez al día en combinación con efavirenz. Delavirdina: La delavirdina tiene el potencial para elevar las concentraciones plasmáticas de lopinavir. Inhibidores de Proteasa (PIs): Amprenavir: Se espera que lopinavir/ritonavir aumente las concentraciones de amprenavir (amprenavir 750 mg dos veces al día más lopinavir/ritonavir produce aumento en el AUC, Cmáx similar, aumento del Cmín, relativo a amprenavir 1200 mg dos veces al día). La co-administración de lopinavir/ritonavir y amprenavir dan lugar a disminución en las concentraciones de lopinavir (ver Dosificación). lopinavir/ritonavir no se debe administrar una vez al día en combinación con amprenavir. Fosamprenavir: Un estudio ha mostrado que la co-administración de lopinavir/ritonavir con fosamprenavir baja las concentraciones de amprenavir y de lopinavir. No se han establecido las dosis apropiadas de la combinación de fosamprenavir y lopinavir/ritonavir con respecto a la seguridad y la eficacia. Indinavir: Se espera que lopinavir/ritonavir aumente las concentraciones de indinavir (indinavir 600 mg dos veces al día más lopinavir/ritonavir produce AUC similar, disminución del Cmáx, aumento del Cmín relativo a indinavir 800 mg tres veces al día. Puede ser necesario disminuir la dosis de indinavir durante la co-administración con lopinavir/ritonavir 400/100 dos veces al día (ver Farmacología: Tabla 10). lopinavir/ritonavir una vez al día no se ha estudiado en combinación con indinavir. Nelfinavir: Se espera que lopinavir/ritonavir aumente las concentraciones de nelfinavir con aumento del metabolito M8 de nelfinavir (1000 mg de nelfinavir dos veces al día más lopinavir/ritonavir produce AUC similar, similar Cmáx, aumento en el Cmín relativo a 1250 mg de nelfinavir dos veces al día). La co-administración de lopinavir/ritonavir y de nelfinavir dan lugar a disminución de las concentraciones de lopinavir (ver Dosis y Administración). Lopinavir/ritonavir no se debe administrar una vez al día en combinación con nelfinavir. Ritonavir: Cuando lopinavir/ritonavir se co-administra con un adicional de ritonavir 100 mg dos veces al día, el AUC de lopinavir aumentó un 33% y Cmín aumentó en 64% comparado con lopinavir/ritonavir 400/100 mg (tres (3) cápsulas blandas de gel) dos veces al día. Saquinavir: Se espera que lopinavir/ritonavir aumente las concentraciones de saquinavir (saquinavir 800 mg dos veces al día más lopinavir/ritonavir produce un aumento en el AUC, aumento en el Cmáx, aumento en el Cmín relativo a 1200 mg de saquinavir tres veces al día). Puede ser necesario disminuir la dosis de saquinavir cuando se co-administra con lopinavir/ritonavir 400/100 mg dos veces al día (ver Farmacología clínica: Tabla 9). lopinavir/ritonavir una vez al día no se ha estudiado en combinación con saquinavir. Otras Drogas: Analgésicos: Fentanyl: Lopinavir/ritonavir inhibe el CYP3A4 y como resultado se espera que aumente las concentraciones plasmáticas de fentanil. Se recomienda un monitoreo cuidadoso de los efectos terapéuticos y adversos (incluyendo depresión respiratoria) cuando se administra fentanilo concomitantemente con lopinavir/ritonavir. Antiarrítmícos (Amiodarona, bepridil, lidocaína sistémica y quinidina): Las concentraciones pueden aumentar cuando se coadministran con lopinavir/ritonavir. Se debe tener precaución y se recomienda monitoreo de la concentración terapéutica cuando está disponible. Digoxina: Un informe de la literatura ha mostrado que la co-administración de ritonavir (300 mg cada 12 horas) y digoxina dio lugar a niveles significativamente aumentados de digoxina. Se debe tener precaución cuando lopinavir/ritonavir se co-administre con digoxina, con la supervisión apropiada de niveles séricos de digoxina Agentes anticancerosos (ej. dasatinib, nilotinib, vincristina, vinblastina): Pueden tener un aumento en sus concentraciones séricas cuando se co-administran con lopinavir/ritonavir produciendo un potencial aumento de los eventos adversos normalmente asociados con estos agentes anticancerosos. Para nilotinib y dasatinib, refiérase a su información prescriptiva para instrucciones de dosificación. Anticoagulantes: Las concentraciones de warfarina pueden ser afectadas cuando se co-administra con lopinavir/ritonavir. Se recomienda que el INR (international normalized ratio) sea monitoreado. Antidepresivos: Bupropión: La administración simultánea de bupropión con lopinavir/ritonavir disminuirá los niveles plasmáticos tanto de bupropión como de su metabolito activo (hidroxibupropión). Trazodona: El uso concomitante de ritonavir y trazodona puede aumentar las concentraciones de trazodona. Se han observado eventos adversos como náuseas, vértigo, hipotensión y síncope. Si se utiliza trazodona con un inhibidor CYP3A4 tal como lopinavir/ritonavir, la combinación se debe utilizar con precaución y se debe considerar una dosis más baja de trazodona. Anticonvulsivantes: (Fenobarbital, fenitoína, carbamazepina): Estas drogas son reconocidas como inductoras de CYP3A4 y pueden disminuir las concentraciones de lopinavir. lopinavir/ritonavir no se debe administrar una vez al día en combinación con fenobarbital, fenitoína o carbamazepina. Además, la co-administración de fenitoína y lopinavir/ritonavir produce un moderado aumento en las concentraciones estado estable de fenitoína. Los niveles de fenitoína se deben monitorear cuando se co-administra con lopinavir/ritonavir. Fungicidas: El ketoconazol y el itraconazol pueden tener concentraciones séricas aumentadas por lopinavir/ritonavir. Dosis altas de ketoconazol e itraconazol (mayor que 200 mg/día) no se recomiendan. Voriconazol: Un estudio ha mostrado que la co-administración de ritonavir 100 mg cada 12 horas disminuyó el AUC estado estable de voriconazol en un promedio de 39%; por lo tanto, la co-administración de lopinavir/ritonavir y voriconazol se debe evitar, a menos que una evaluación del beneficio/riesgo para el paciente justifique el uso de voriconazol. Antibióticos: Se esperan moderados incrementos en el AUC de claritromicina cuando se co-administra con lopinavir/ritonavir. Para los pacientes con daño hepático o renal se debe considerar disminución de la dosis de claritromicina. Antimicobacterianos: Cuando la rifabutina y lopinavir/ritonavir son co-administrados por 10 días, la rifabutina (droga madre y el metabolito activo 25-0-desacetilo), el Cmáx y AUC se incrementan 3.5 y 5.7 veces, respectivamente. Sobre la base de estos datos, una reducción en la dosis de rifabutina de 75% (ej. 150 mg día por medio o 3 veces por semana) se recomienda cuando se administra con lopinavir/ritonavir. Además, puede ser necesaria una reducción de la dosis de rifabutina. Debido a grandes disminuciones en las concentraciones de lopinavir, la rifampicina no debe ser usado en combinación con la dosis estándar de lopinavir/ritonavir (ver Advertencias: Interacciones de Droga). El uso de Rifampicina con la dosis estándar de lopinavir/ritonavir, puede conducir a la pérdida de respuesta virológica y a posible resistencia a lopinavir/ritonavir o a la clase de inhibidores de proteasa o a otros agentes antiretrovirales co-administrados. La co-administración de rifampicina con 800/200 mg lopinavir/ritonavir dos veces al día produjo disminuciones en lopinavir de hasta 57% y con lopinavir/ritonavir 400/400 mg dos veces al día produjo disminuciones de hasta 7% cuando se comparó con lopinavir/ritonavir 400/100 mg dos veces al día dosificado en ausencia de rifampicina. Elevaciones en ALT y AST se han notado en estudios con dosis mayores de lopinavir/ritonavir co-administrado con rifampicina y puede ser dependiente a la secuencia de administración de dosis. Si se está considerando la co-administración, lopinavir/ritonavir se debe iniciar en dosis estándar por aproximadamente 10 días previos a la adición de rifampicina. Luego, la dosis de lopinavir/ritonavir se debe titular hacia arriba. Se indica un monitoreo muy cercano de la función hepática (ver Farmacología para magnitud de interacción, Tabla 9). Antiparasitarios: Es posible que disminuya la concentración terapéutica de atovacuona cuando se co-administra con lopinavir/ritonavir. Aumentar las dosis de atovacuona puede ser necesario. Corticoides: La dexametasona puede inducir CYP3A4 y puede disminuir las concentraciones de lopinavir. Propionato de Fluticasona: El uso concomitante de propionato de Fluticasona y lopinavir/ritonavir puede aumentar las concentraciones de propionato de fluticasona. Use con precaución. Considere las alternativas para propionato de fluticasona, particularmente para el uso a largo plazo (ver Advertencias: Interacciones de Droga). Dihidropiridinas Bloqueadores de los canales del calcio: (ej. Felodipino, nifedipino, nicardipino): Pueden aumentar sus concentraciones séricas por lopinavir/ritonavir. Agentes para la disfunción eréctil (inhibidores PDE5): Sildenafil: Para el tratamiento de la disfunción eréctil, usar sildenafil con precaución y en dosis reducidas de 25 mg cada 48 hrs. con aumento del monitoreo de eventos adversos (ver Advertencias: Interacciones de Droga). El uso concomitante de sildenafil con lopinavir/ritonavir está contraindicado en pacientes con hipertensión arterial pulmonar (ver Contraindicaciones). Tadalafil: usar tadalafil con precaución en dosis reducidas de no más de 10 mg cada 72 horas con aumento en el monitoreo de eventos adversos (ver Advertencias: Interacciones de Droga). Vardenafil: usar vardenafil con precaución en dosis reducidas de no más de 2.5 mg cada 72 horas con aumento en el monitoreo de eventos adversos (ver Advertencias: Interacciones de Droga). Productos herbales: Pacientes en tratamiento con lopinavir/ritonavir no deben usar productos que contengan Hierba de San Juan concomitantemente, ya que esta combinación puede provocar concentraciones plasmáticas reducidas de lopinavir/ritonavir. Este efecto se puede deber a una inducción de CYP3A4 y puede ocasionar pérdida del efecto terapéutico y desarrollo de resistencia (ver Contraindicaciones y Advertencias: Interacciones de Droga). Inhibidores de la HMG-CoA reductasa: Los inhibidores de la HMG-CoA reductasa son altamente dependientes del metabolismo de CYP3A4, tales como la lovastatina y simvastatina, se espera que tengan marcados aumentos de las concentraciones plasmáticas cuando se co-administran con lopinavir/ritonavir. Ya que las concentraciones aumentadas de inhibidores de la HMG-CoA reductasa pueden causar miopatía, incluyendo rabdomiólisis, la combinación de estas drogas con lopinavir/ritonavir está contraindicada (ver Contraindicaciones). La atorvastatina es menos dependiente del CYP3A para el metabolismo. Cuando la atorvastatina fue administrada concomitantemente con lopinavir/ritonavir, se observó un aumento medio de 4,7 y 5,9 veces en el Cmáx y AUC de la atorvastatina, respectivamente. Cuando se use con lopinavir/ritonavir, se debe administrar la dosis más baja posible de atorvastatina. Los resultados de un estudio de interacción de droga con lopinavir/ritonavir y pravastatina no revelaron interacción clínica significativa. El metab
Conservación.
Almacenar a no más de 25°C.
Sobredosificación.
La experiencia de sobredosis aguda con lopinavir/ritonavir en seres humanos es limitada. El tratamiento de la sobredosis con lopinavir/ritonavir debe consistir en medidas generales de apoyo, incluyendo monitoreo de los signos vitales y observación del estado clínico del paciente. No existe ningún antídoto específico para la sobredosis con lopinavir/ritonavir. Si estuviera indicado, se deberá proceder a la eliminación de la droga no absorbida mediante emesis o lavado gástrico. Para ayudar a eliminar la droga no absorbida también se puede administrar carbón activado. Debido a que lopinavir/ritonavir se une extensamente a las proteínas, es poco probable que la diálisis sea de beneficio en la extracción de una porción significativa de la droga.
Presentación.
Comprimidos recubiertos 200/50 mg: Los comprimidos recubiertos de Lopinavir/ritonavir son amarillos, con una cobertura realzada con la insignia Abbott y el Código KA. Lopinavir/ritonavir está disponible como comprimido recubierto de 200 mg de lopinavir/50 mg de ritonavir.


 Cambiar de país
Cambiar de país