CEUMID
MEGALABS
Anticonvulsivante.
Composición.
CEUMID 500: Cada comprimido contiene levetiracetam 500 mg. CEUMID 1000: Cada comprimido contiene levetiracetam 1000 mg. Excipientes: Polividona, almidón de maíz, colorante FD&C rojo N° 40 laca alumínica (CI N° 16035), dióxido de silicio coloidal, talco, estearato de magnesio, hipromelosa, macrogol 6000, dióxido de titanio (CI N° 77891). CEUMID solución oral 100 mg/mL: Cada mL de solución contiene: Levetiracetam 100 mg. Excipientes: Metilparabeno, propilparabeno, citrato de sodio dihidratado, ácido cítrico anhidro, glicerina, sorbitol solución, acesulfamo potásico, esencia de ananá, enmascarante de sabor, agua purificada c.s. CEUMID IV 500 mg: Cada mL de concentrado para solución para perfusión contiene 100 mg de Levetiracetam. Cada vial de 5 mL contiene 500 mg de Levetiracetam; Excipientes: cloruro de sodio, acetato de sodio trihidrato, ácido acético glacial, agua para inyección, c.s.
Propiedades.
Mecanismo de acción: Se desconocen los mecanismos concretos mediante los cuales CEUMID ejerce su efecto antiepiléptico, pero no parecen derivar de ninguna interacción con mecanismos conocidos que participan en la neurotransmisión inhibitoria y excitatoria. Las experienctas in vitro e in vivo sugieren que CEUMID no altera las características celulares básicas ni la neurotransmisión normal. Estudios in vitro muestran que levetiracetam afecta los niveles intraneuronales de Ca2+ mediante inhibición parcial de las corrientes de Ca2+ tipo N, reduciendo la liberación de Ca2+ de la reserva intraneuronal. Además, invierte parcialmente la reducción de corrientes dependientes de GABA y glicina inducidas por zinc y ß-carbolinas. Por otra parte, estudios in vitro muestran que levetiracetam se une a la proteína 2A de las vesículas sinápticas, lo cual parece estar involucrado en la fusión de vesículas y en la exocitosis de neurotransmisores. Levetiracetam y sus análogos han mostrado un orden de afinidad por la unión a la proteína 2A de las vesículas sinápticas que se correlaciona con la potencia de la protección contra los ataques epilépticos en el modelo audiogénico de epilepsia en ratón. Este hallazgo sugiere que la interacción entre levetiracetam y la proteína 2A de las vesículas sinápticas parece contribuir en el mecanismo de acción del fármaco como antiepiléptico.
Farmacocinética.
Adultos y adolescentes: Absorción: Levetiracetam se absorbe rápidamente después de su administración oral. La biodisponibilidad oral absoluta es cercana al 100%. El peak de nivel plasmático (Cmax) se alcanza a las 1,3 horas de su administración. Los niveles plasmáticos estables se obtienen a los dos días con la pauta de administración de dos veces al día. Los valores normales del peak plasmático (Cmax) después de una dosis simple de 1.000 mg y de una dosis repetida de 1.000 mg dos veces al día son del 31 y 43 mg/ml respectivamente. El grado de absorción es dosis-independiente y no está alterado por los alimentos. Distribución: No se dispone de datos de distribución tisular en humanos. Ni levetiracetam ni su metabolito primario se unen de forma significativa a las proteínas plasmáticas ( < 10%). El volumen de distribución del levetiracetam es aproximadamente de 0,5 a 0,7 l/kg, valor cercano al volumen total del agua corporal. Biotransformación: Levetiracetam no se metaboliza extensamente en humanos. La vía metabólica principal (24% de la dosis) es la hidrólisis enzimática del grupo acetamida. La formación del metabolito primario, ucb L057, no depende de ninguna de las isoformas del citocromo P450 hepático. La hidrólisis del grupo acetamida fue mesurable en un gran número de tejidos, incluyendo las células sanguíneas. El metabolito ucb L057 es farmacológicamente inactivo. Se identificaron también dos metabolitos minoritarios. Uno estaba formado por la hidroxilación del anillo de la pirrolidona (1,6% de la dosis) y el otro por la apertura del anillo de la pirrolidona (0,9 % de la dosis). Otros compuestos no identificados representaban solamente el 0,6% de la dosis. No se evidenció interconversión enantiomérica in vivo para levetiracetam o para su metabolito primario. Los estudios in vitro han mostrado que levetiracetam y su metabolito principal no inhiben las isoformas principales del citocromo P450 hepático humano (CYP3A4, 2A6, 2C9, 2C19, 2D6, 2E1 y 1A2), la glucuronil transferasa (UGT1A1 y UGT1A6) y la actividad de la epóxido hidroxilasa. Además, levetiracetam no afecta la glucuronidación in vitro del ácido valproico. En cultivos de hepatocitos humanos, levetiracetam tuvo poco o ningún efecto sobre el CYP1A2, SULT1E1 o UGT1A1. Levetiracetam provocó una leve inducción del CYP2B6 y del CYP3A4. Los datos de interacciones in vitro e in vivo con anticonceptivos orales, digoxina y warfarina indican que no se espera que exista una inducción enzimática significativa in vivo. Por consiguiente, es muy poco probable que levetiracetam interaccione con otras sustancias, o viceversa. Eliminación: La vida media plasmática en adultos fue de 7 a 8 horas y no varía con la dosis, con la vía de administración o con la administración repetida. El aclaramiento corporal total medio fue de 0,96 ml/min/kg. La vía de excreción principal fue por la urinaria, alcanzando una media del 95% de la dosis (aproximadamente un 93 % de la dosis se excretaba dentro de las primeras 48 horas). La excreción por vía fecal representaba solamente el 0,3 % de la dosis. La excreción urinaria acumulada de levetiracetam y de su metabolito primario durante las primeras 48 horas alcanzó, respectivamente, el 66% y el 24% de la dosis. El aclaramiento renal de levetiracetam y de ucb L057 es de 0,6 y de 4,2 ml/min/kg respectivamente, lo que indica que levetiracetam se excreta por filtración glomerular con subsiguiente reabsorción tubular y que el metabolito primario se excreta también por secreción tubular activa en adición a la filtración glomerular. La eliminación de levetiracetam está correlacionada con el aclaramiento de creatinina. Pacientes de edad avanzada: En la vejez, la vida media se incrementa alrededor de un 40% (10 a 11 horas). Esto está relacionado con la disminución de la función renal en esta población. Insuficiencia renal: El aclaramiento corporal aparente está correlacionado con el aclaramiento de creatinina, tanto para levetiracetam como para su metabolito primario. Así, en pacientes con insuficiencia renal moderada o grave se recomienda ajustar la dosis diaria de mantenimiento de levetiracetam en base al aclaramiento de creatinina. En sujetos adultos con patología renal terminal anúrica la vida media fue aproximadamente de 25 y de 3,1 horas durante los períodos interdiálisis e intradiálisis respectivamente. La fracción de levetiracetam eliminada durante una sesión de diálisis normal de 4 horas fue de un 51%. Insuficiencia hepática: En sujetos con insuficiencia hepática leve o moderada no hubo modificación relevante del aclaramiento de levetiracetam. En la mayoría de los sujetos con insuficiencia hepática grave el aclaramiento de levetiracetam se redujo en más del 50% como consecuencia de la insuficiencia renal concomitante. Población pediátrica: Niños (de 4 a 12 años): Después de la administración de una dosis oral única (20 mg/kg) a niños epilépticos (de 4 a 12 años), la vida media de levetiracetam fue de 6 horas. El aclaramiento corporal aparente ajustado al peso fue alrededor de un 30% más alto que en los adultos epilépticos. Tras la administración de dosis orales repetidas (de 20 a 60 mg/kg/día) a niños epilépticos (de 4 a 12 años), levetiracetam se absorbió rápidamente. El peak de concentración plasmática se observó entre 0,5 y 1 hora después de la administración. Se observaron incrementos lineales y proporcionales a la dosis para los peaks de concentraciones plasmáticas y del área bajo la curva. La vida media de eliminación fue aproximadamente de 5 horas. El aclaramiento corporal aparente fue de 1,12 mL/min/kg. En el análisis farmacocinético poblacional realizado en pacientes desde 1 mes a 16 años de edad, el peso corporal estuvo significativamente relacionado con el aclaramiento aparente (aumento en el aclaramiento con aumento del peso corporal) y con el volumen de distribución aparente. La edad también tuvo influencia sobre ambos parámetros. Este efecto fue pronunciado para los lactantes más pequeños y decreció al ir aumentando la edad hasta hacerse insignificante alrededor de los 4 años de edad. En ambos análisis farmacocinéticos poblacionales hubo alrededor de un 20% de aumento en el aclaramiento aparente de levetiracetam cuando se administró conjuntamente con un medicamento antiepiléptico inductor enzimático. No se ha investigado la farmacocinética tras la administración intravenosa en pacientes pediátricos. Sin embargo, basándonos en las características famacocinéticas de Levetiracetam tras la administración intravenosa en adultos y en la farmacocinética tras la administración oral en niños, se espera que la exposición (AUC) a Levetiracetam sea similar en pacientes pediátricos de 4 a 12 años tras la administración oral y tras la administración intravenosa. Una dosis única de 1.500 mg de levetiracetam diluida en 100 mL de un diluyente compatible y administrada por perfusión intravenosa durante 15 minutos es bioequivalente a una toma por vía oral 1.500 mg de Levetiracetam, administrada en 3 comprimidos de 500 mg. Se evaluó la administración intravenosa de dosis de hasta 4.000 mg diluidas en 100 mL de cloruro de sodio al 0,9 % perfundidas durante 15 minutos y dosis de hasta 2.500 mg diluidas en 100 mL de cloruro de sodio al 0,9 % perfundidas durante 5 minutos. Los perfiles farmacocinéticos y de seguridad no identificaron ningún aspecto problemático relacionado con la seguridad. Levetiracetam es un compuesto muy soluble y permeable. El perfil farmacocinético es lineal y con poca variabilidad intra e interindividual. No hay modificación del aclaramiento después de la administración repetida. El perfil farmacocinético no dependiente del tiempo de Levetiracetam también se determinó mediante la perfusión intravenosa de una dosis de 1.500 mg durante 4 días administrada dos veces al día. No hay evidencia de variabilidad relevante de género, raza o circadiana. El perfil farmacocinético en voluntarios sanos y en pacientes con epilepsia es comparable.
Indicaciones.
CEUMID, está indicado como monoterapia en el tratamiento de la crisis epiléptica de inicio parcial, con o sin generalización secundaria, en pacientes mayores de 16 años con un nuevo diagnóstico de epilepsia. CEUMID está indicado como terapia concomitante en: El tratamiento de las crisis de inicio parcial con o sin generalización secundaria en adultos y niños mayores de 4 años con epilepsia. El tratamiento de las crisis mioclónicas en adultos y adolescentes mayores de 12 años con epilepsia mioclónica juvenil. El tratamiento de las crisis tónico-clónicas generalizadas primarias en adultos y adolescentes mayores de 12 años con epilepsia idiopática generalizada.
Dosificación.
Vía oral: Los comprimidos recubiertos deben ser tomados en forma oral, y tragados con una cantidad suficiente de líquido y pueden ser tomados con o sin alimentos. La dosis recomendada diaria son 2 dosis equitativas repartidas dos veces al día. La solución oral puede diluirse en un vaso de agua o en una mamadera y puede administrarse con o sin alimentos. Junto con la solución oral de CEUMID se incluye una copa dosificadora graduada. La posología diaria se divide en dosis iguales repartidas en dos tomas al día. Una dosis de 100 mg equivale a 1 mL del jarabe. Monoterapia: Dosis en adultos y adolescentes (desde 16 años de edad): CEUMID comprimidos: La dosis inicial recomendada es 250 mg dos veces al día, la cual debe incrementarse hasta la dosis terapéutica inicial de 500 mg dos veces al día tras dos semanas de tratamiento. La dosis puede incrementarse en 250 mg dos veces al día cada dos semanas dependiendo de la respuesta al tratamiento. La dosis máxima corresponde a 1.500 mg dos veces al día. CEUMID solución: Al inicio del tratamiento se recomienda una dosis baja (250 mg = 2,5 mL, en dos tomas al día), durante dos semanas antes de administrarse la dosis de mantenimiento (entre 1.000 mg y 3.000 mg al día equivalentes a 10 y 30 mL, respectivamente). La dosis se ira ajustando según cada caso en particular. La modificación de la dosis se puede realizar con aumentos o reducciones de 500 mg (5 mL) dos veces al día cada dos a cuatro semanas. CEUMID IV: La dosis inicial recomendada es de 250 mg dos veces al día, la cual debe aumentarse hasta la dosis terapéutica inicial de 500 mg dos veces al día tras dos semanas de tratamiento. La dosis puede aumentarse en función de la respuesta clínica con CEUMID IV 250 mg dos veces al día cada 2 semanas. La dosis máxima de CEUMID IV es de 1.500 mg dos veces al día. Límite de prescripción en adultos al día: 3.000 mg. No se ha establecido la seguridad y eficacia como monoterapia en niños y adolescentes menores de 16 años. Terapia concomitante: Dosis en niños (de 4 a 11 años) y adolescentes (de 12 a 17 años) con un peso inferior a 50 kg. CEUMID comprimidos: La dosis terapéutica inicial es de 500 mg dos veces al día. Esta dosis se puede instaurar desde el primer día de tratamiento. Dependiendo de la respuesta clínica y de la tolerabilidad, la dosis diaria se puede incrementar hasta 1.500 mg dos veces al día. La modificación de la dosis se puede realizar con aumentos o reducciones de 500 mg dos veces al día cada dos a cuatro semanas. CEUMID solución: La dosis terapéutica inicial es de 10 mg/kg dos veces al día. En función de la respuesta clínica y de la tolerabilidad, se puede aumentar la dosis hasta los 30 mg/kg dos veces al día. Los cambios de dosis no deben exceder de aumentos o reducciones de 10 mg/kg dos veces al día cada dos semanas. Se debe utilizar la dosis menor eficaz. La dosis en niños con un peso de 50 kg o superior es la misma que en adultos. Niños con más de 20 kg, pueden ser tratados con comprimidos o solución oral. Utilizar la siguiente fórmula para establecer la dosis diaria en pacientes pediátricos con CEUMID solución oral, sobre la base de una dosis diaria de 20 mg/kg/día, 40 mg/kg/día o 60 mg/kg/día: Dosis diaria total (día) = Dosis diaria (mg/kg/día) x peso corporal (kg)/100 mg/mL. Uso en niños menores de 4 años: levetiracetam no está recomendado en este grupo de la población al no haber datos suficientes sobre su seguridad y eficacia. CEUMID IV: La dosis terapéutica inicial es de 10 mg/kg dos veces al día. En función de la respuesta clínica y de la tolerabilidad, se puede aumentar la dosis hasta los 30 mg/kg dos veces al día. Los cambios de dosis no deben exceder de aumentos o reducciones de 10 mg/kg dos veces al día cada dos semanas. Se debe utilizar la dosis menor eficaz. La dosis en niños con un peso de 50 kg o superior es la misma que en adultos. 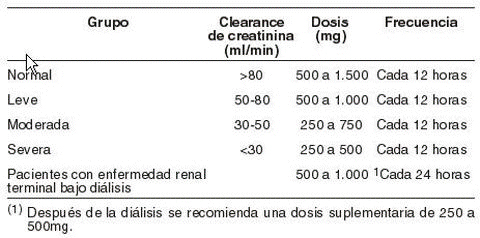
Uso en niños menores de 4 años de edad: Levetiracetam concentrado para solución para perfusión 500 mg/5 mL no está recomendado para usarlo en niños menores de 4 años debido a la escasez de datos de seguridad y eficacia en esta población. Dosis en adultos (≥18 años) y adolescentes (de 12 a 17 años) con un peso de 50 kg o superior: La dosis terapéutica inicial es de 500 mg (5 mL) dos veces al día. Esta dosis se puede instaurar desde el primer día de tratamiento. Dependiendo de la respuesta clínica y de la tolerabilidad, la dosis diaria se puede incrementar hasta 1.500 mg (15 mL) dos veces al día. La modificación de la dosis se puede realizar con aumentos o reducciones de 500 mg (5 mL) dos veces al día cada dos a cuatro semanas. Insuficiencia renal: La dosis diaria se debe individualizar de acuerdo con la función renal. Para pacientes adultos, referirse a la siguiente tabla y ajustar la dosis indicada. Para utilizar esta tabla de dosificación se necesita una estimación del aclaramiento de creatinina (CLcr), en ml/min, del paciente.El CLcr, en ml/min, se puede estimar a partir de la determinación de la creatinina sérica (mg/dl) para adultos y adolescentes que pesen 50 kg o más utilizando la siguiente fórmula: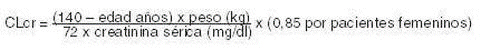
Ajuste de dosis para pacientes adultos y adolescentes que pesan más de 50 kg, con función renal alterada:
Para niños con insuficiencia renal, la dosis de Levetiracetam necesita ser ajustada en base a la función renal, ya que la depuración de levetiracetam está relacionada con la función renal. Esta recomendación está basada en un estudio en pacientes adultos con insuficiencia renal. La CLcr en mL/min/1,73 m2 puede estimarse a partir de la determinación de creatinina sérica (mg/dL) utilizando la siguiente fórmula para adolescentes jóvenes y niños (fórmula Schwartz):
Ajuste de dosis para pacientes niños y adolescentes que pesan menos de 50 kg con función renal alterada:
Insuficiencia hepática: No es necesario ajustar la dosis en pacientes con insuficiencia hepática leve a moderada. En pacientes con insuficiencia hepática grave, el aclaramiento de creatinina puede subestimar el grado de insuficiencia renal. Por lo tanto, se recomienda una reducción del 50% de la dosis de mantenimiento diario cuando el aclaramiento de creatinina es < 60 ml/min/1,73 m2. Forma de administración: El tratamiento con CEUMID puede iniciarse tanto por administración intravenosa como por administración oral. La conversión desde la administración oral a la administración intravenosa y viceversa, puede hacerse directamente sin modificar la dosis. Debería mantenerse la dosis diaria total y la frecuencia de administración. CEUMID IV concentrado es solamente para uso por vía intravenosa y la dosis recomendada debe diluirse como mínimo en 100 ml de un diluyente compatible y administrarse por vía intravenosa como una perfusión intravenosa de 15 minutos. Diluyentes: Inyección de cloruro de sodio (0,9%). Inyección de Ringer lactato. Inyección de Dextrosa 5%.
Contraindicaciones.
CEUMID, no debe utilizarse en casos de conocida hipersensibilidad a levetiracetam, u otros derivados de la pirrolidona o a cualquiera de los componentes de la fórmula.
Reacciones adversas.
Datos de Ensayos Clínicos y Datos Post-Comercialización: Resumen del perfil de seguridad: El perfil de eventos adversos presentado a continuación está basado en el análisis de estudios clínicos comparativos con placebo acumulados con todas las indicaciones estudiadas, con un total de 3.416 pacientes tratados con levetiracetam. Estos datos son complementados con el uso de levetiracetam en estudios de extensión sin anonimato correspondientes, así como con la experiencia post-comercialización. Las reacciones adversas más frecuentemente reportadas fueron nasofaringitis, somnolencia, cefalea, fatiga y mareo. El perfil de seguridad de levetiracetam es generalmente similar entre los grupos de edad (pacientes adultos y pediátricos) y entre las indicaciones para epilepsia aprobadas. Las reacciones adversas al medicamento (ADR por su sigla en inglés) están enlistadas bajo el sistema de clasificación de órganos MedDRA y frecuencia. Las frecuencias están definidas como: Muy común: ≥1/10. Común: ≥1/100 a < 1/10. Poco común: ≥1/1.000 a < 1/100. Rara: ≥1/10.000 a < 1/1.000. Muy rara: < 1/10.000. Desconocida: no se puede estimar a partir de los datos disponibles. Infecciones e infestaciones: Muy común: nasofaringitis. Rara: infección. Trastornos de la sangre y del sistema linfático: Poco común: trombocitopenia, leucopenia. Rara: pancitopenia, neutropenia, agranulocitosis. Trastornos del sistema inmune: Rara: reacción farmacológica con eosinofilia y síntomas sistémicos (DRESS por sus siglas en inglés), hipersensibilidad (incluyendo angioedema y anafilaxia). Trastornos del metabolismo y de la nutrición: Común: anorexia. Poco común: peso disminuido, aumento de peso. Rara: hiponatremia. Trastornos psiquiátricos: Común: depresión, hostilidad/agresión, ansiedad, insomnio, nerviosismo/irritabilidad. Poco común: intento de suicidio, ideación suicida, trastorno psicótico, comportamiento anormal, alucinación, reacción de ira, estado confusional, labilidad afectiva/cambios del estado de ánimo, agitación. Rara: suicidio consumado, trastorno de la personalidad, pensamiento anormal. Trastornos del sistema nervioso: Muy común: somnolencia, cefalea. Común: convulsión, trastorno del equilibrio, mareo, letargo, temblor. Poco común: amnesia, alteración de la memoria, coordinación anormal/ataxia, parestesia, alteración de la atención. Rara: coreoatetosis, discinesia, hiperquinesia. Trastornos oculares: Poco común: diplopía, visión borrosa. Trastornos del oído y del laberinto: Común: vértigo. Trastornos respiratorios, torácicos y mediastínicos. Común: tos. Trastornos gastrointestinales: Común: dolor abdominal, diarrea, dispepsia, vómito, náusea. Rara: pancreatitis. Trastornos hepatobiliares: Poco común: Prueba anormal de función hepática. Rara: Insuficiencia hepática, hepatitis. Trastornos de la piel y del tejido subcutáneo: Común: erupción. Poco común: alopecia, eczema, prurito. Rara: necrólisis epidérmica tóxica, Síndrome de Stevens-Johnson, eritema multiforme. Trastornos musculoesqueléticos y del tejido conjuntivo: Poco común: debilidad muscular, mialgia. Trastornos generales y alteraciones en el lugar de administración: Común: astenia, fatiga. Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos: Poco común: lesión. Descripción de reacciones adversas seleccionadas: El riesgo de anorexia es mayor cuando levetiracetam es co-administrado con topiramato. En algunos casos de alopecia, se observó la recuperación cuando se descontinuó levetiracetam. Se identificó supresión de médula ósea en algunos de los casos de pancitopenia. Población pediátrica: En pacientes con edad de 1 mes a menos de 4 años de edad, un total de 190 pacientes habían sido tratados con levetiracetam en estudios comparativos con placebo y sin anonimato. Sesenta de estos pacientes se trataron con levetiracetam en estudios comparativos con placebo. En pacientes de 4 a 16 años de edad, un total de 645 pacientes han sido tratados con levetiracetam en los ensayos controlados con placebo y de extensión abierta. De estos, 233 pacientes fueron tratados con levetiracetam en los ensayos controlados con placebo. En ambos rangos de edad pediátricos, estos datos se complementan con la experiencia post-comercialización del uso de levetiracetam. Además, 101 lactantes menores de 12 meses han sido expuestos en un estudio de seguridad post autorización. No se identificaron nuevas preocupaciones acerca de la seguridad por levetiracetam para lactantes menores de 12 meses de edad con epilepsia. El perfil de seguridad de levetiracetam es, en general, similar en todos los grupos de edad y en todas las indicaciones aprobadas en epilepsia. Los resultados de seguridad de los ensayos clínicos controlados con placebo en pacientes pediátricos coincidieron con el perfil de seguridad de levetiracetam en adultos excepto por las reacciones adversas psiquiátricas y de comportamiento, las cuales fueron más frecuentes en niños que en adultos. En niños y adolescentes de 4 a 16 años de edad, vómitos (muy comunes, 11,2%), agitación (común, 3,4%), cambios de humor (común, 2,1%), inestabilidad emocional (común, 1,7%), agresividad (común, 8,2%), comportamiento anormal (común, 5,6%) y letargo (común, 3,9%) fueron notificados más frecuentemente que otros rangos de edad o que en el perfil de seguridad global. En lactantes y niños de 1 mes a menos de 4 años de edad, irritabilidad (muy común, 11,7%) y coordinación anormal (común, 3,3%) fueron notificados más frecuentemente que en otros grupos de edad o que en el perfil de seguridad global. Un estudio de seguridad pediátrica doble ciego controlado con placebo con diseño de no-inferioridad ha evaluado los efectos cognitivos y neuropsicológicos de Levetiracetam en niños de 4 a 16 años de edad con crisis de inicio parcial. Se concluyó que Levetiracetam no era diferente (no era inferior) a placebo con respecto al cambio en la puntación en la escala "Leiter-R Attention and Memory, Memory Screen Composite" desde el inicio en la población por protocolo. Los resultados relacionados con la función emocional y el comportamiento, medidos de forma estandarizada y sistemática usando un instrumento validado (cuestionario CBCL de Achenbach), indicaron un empeoramiento del comportamiento agresivo en los pacientes tratados con Levetiracetam. Sin embargo, los sujetos que tomaron Levetiracetam en el ensayo de seguimiento a largo plazo, abierto, no experimentaron un empeoramiento, en promedio, en su función emocional y comportamiento; en concreto, las medidas del comportamiento agresivo no empeoraron con respecto al inicio.
Precauciones.
Supresión del tratamiento: De acuerdo con las recomendaciones habituales, si se ha de suprimir la medicación con CEUMID se recomienda retirarlo de forma gradual (en adultos y adolescentes que pesen más de 50 kg, conviene hacer reducciones de 500 mg dos veces al día cada dos a cuatro semanas; en niños y adolescentes que pesen menos de 50 kg, las reducciones de dosis no deberían exceder de los 10 mg/kg dos veces al día, cada dos semanas). Insuficiencia renal y hepática: La administración de levetiracetam a pacientes con insuficiencia renal puede requerir el ajuste de la dosis. En pacientes con insuficiencia hepática grave se recomienda valorar la función renal antes de la selección de la dosis. Suicidio: Con el uso de fármacos antiepilépticos (incluyendo levetiracetam) se han notificado casos de suicidio, intento de suicidio, pensamientos y comportamientos suicidas en pacientes tratados con fármacos antiepilépticos (incluyendo levetiracetam). Un metaanálisis de ensayos controlados con placebo, aleatorizados, con fármacos antiepilépticos ha mostrado un pequeño aumento del riesgo de pensamientos y comportamientos suicidas. Se desconoce el mecanismo de este riesgo. Por tanto, los pacientes bajo tratamiento con anticolvulsivantes deben ser monitorizados para detectar signos de depresión y/o pensamientos y comportamientos suicidas y debe considerarse el tratamiento adecuado. Se debe aconsejar a los pacientes (y a sus cuidadores) que consulten con el médico tratante si aparecen signos de depresión y/o pensamientos suicidas. Reacciones psiquiátricas: En algunos pacientes levetiracetam puede provocar alteraciones en la conducta. La incidencias de anomalías de comportamiento en los estudios de las convulsiones tónico- clónicas generalizadas y mioclónicas primaria fueron comparables a los de los estudios de las convulsiones de inicio parcial adultos y pediátricos. Cerca del doble de pacientes tratados con levetiracetam adultos y pediátricos (de 4 a 16 años) en comparación con los pacientes adultos y pediátricos tratados con placebo, presentaron síntomas conductuales no psicóticos (reportados como agresión, agitación, ira, ansiedad, apatía, despersonalización, depresión, labilidad emocional, hostilidad, hipercinesia, irritabilidad, nerviosismo, neurosis y trastornos de la personalidad). Población pediátrica: Los datos disponibles en niños no sugieren ningún efecto en el crecimiento ni en la pubertad. No obstante, siguen sin conocerse los efectos a largo plazo sobre el aprendizaje, inteligencia, crecimiento, función endocrina, pubertad y fertilidad en niños. Reacciones dermatológicas graves: Se han reportado en niños y adultos tratados con levetiracetam reacciones dermatológicas graves, incluyendo el síndrome de Stevens-Johnson (SSJ) y necrólisis epidérmica tóxica (NET). El tiempo medio de aparición es de 14 a 17 días, pero se han reportado casos al menos cuatro meses después de iniciar el tratamiento. También se ha informado recurrencia de las reacciones de la piel graves tras la reexposición con levetiracetam. Ante la primera señal de una erupción debe suspenderse levetiracetam, a menos que la erupción no esté claramente relacionada con fármacos. Si los signos o síntomas sugieren SSJ/NET, el uso de este medicamento no debe reanudarse y debe ser considerado un tratamiento alternativo. Alteraciones hematológicas: Se han reportado conteos ligeramente menores de los glóbulos rojos, hemoglobina y hematocrito con respecto a la serie roja. También se han reportado disminuciones estadísticamente significativas en los leucocitos, linfocitos y neutrofilos. Embarazo y la lactancia (salvo indicación médica en contrario). Embarazo: Los estudios en animales han mostrado toxicidad reproductiva. Levetiracetam no se recomienda durante el embarazo ni en mujeres en edad fértil que no utilicen métodos anticonceptivos efectivos, a menos que sea clínicamente necesario. Al igual que con otros medicamentos antiepilépticos, los cambios fisiológicos durante el embarazo pueden afectar las concentraciones de levetiracetam. Se ha observado la disminución de las concentraciones plasmáticas de levetiracetam durante el embarazo. Esta disminución es más pronunciada durante el tercer trimestre (hasta el 60% de la concentración inicial antes del embarazo). Debe asegurarse un control clínico adecuado de la mujer embarazada tratada con levetiracetam. La retirada de los tratamientos antiepilépticos puede dar lugar a una exacerbación de la enfermedad, que podría perjudicar a la madre y al feto. Lactancia: Levetiracetam se excreta en la leche materna humana, por lo que no se recomienda la lactancia natural. Sin embargo, si durante el periodo de lactancia es necesario el tratamiento con levetiracetam, debe considerarse la relación beneficio/riesgo del tratamiento teniéndose en cuenta la importancia de la lactancia natural. Efectos sobre la capacidad para conducir y utilizar máquinas: No se han realizado estudios de los efectos sobre la capacidad para conducir y utilizar máquinas. Debido a las posibles diferencias de sensibilidad individual algunos pacientes pueden experimentar somnolencia, fatiga, alteraciones en la coordinación u otros síntomas relacionados con el sistema nervioso central, especialmente al inicio del tratamiento (primeras 4 semanas del tratamiento) o después de un incremento de la dosis. Por tanto, se recomienda precaución a los pacientes cuando realicen tareas que requieran habilidad específica, p. ej. conducir vehículos o utilizar maquinaria. Se aconseja a los pacientes no conducir o utilizar maquinaria hasta que se compruebe que su capacidad para realizar estas actividades no queda afectada.
Interacciones.
Los datos de los estudios clínicos, realizados en adultos antes de la comercialización, indican que levetiracetam no influye en las concentraciones séricas de los medicamentos antiepilépticos conocidos (fenitoína, carbamazepina, ácido valproico, fenobarbital, lamotrigina, gabapentina y primidona) y que estos medicamentos antiepilépticos no influyen en la farmacocinética de levetiracetam. Como en adultos, en pacientes pediátricos no hay una evidencia clara de interacciones farmacológicas clínicamente significativas, en aquellos que hayan tomado hasta 60 mg/kg/día de levetiracetam. Probenecid, Se ha comprobado que probenecid (500 mg cuatro veces al día), agente bloqueante de la secreción tubular renal, inhibe el aclaramiento renal del metabolito primario pero no el de levetiracetam. Con todo, los niveles de este metabolito se mantienen bajos. Es de esperar que otros fármacos que se excretan por secreción tubular activa puedan reducir también el aclaramiento renal del metabolito. No se ha estudiado el efecto del levetiracetam sobre el probenecid y no se conoce el efecto de levetiracetam sobre otros fármacos secretados activamente, p. ej. AINES, sulfonamidas y metotrexato. Otras interacciones farmacocinéticas: Dosis diarias de 1.000 mg de levetiracetam no influenciaron la farmacocinética de los anticonceptivos orales (etinilestradiol y levonorgestrel); no se modificaron los parámetros endocrinos (hormona luteinizante y progesterona). Dosis diarias de 2.000 mg de levetiracetam no influenciaron la farmacocinética de la digoxina y de la warfarina; no se modificó el tiempo de protrombina. La coadministración con digoxina, anticonceptivos orales y warfarina no tuvo influencia sobre la farmacocinética del levetiracetam. Alcohol: No se dispone de datos sobre la interacción del levetiracetam con alcohol. Incompatibilidades: Este medicamento no debe mezclarse con otros medicamentos excepto con los siguientes (mencionados también en la sección de dosis y forma de administración): Diluyentes: Inyección de cloruro de sodio (0,9%). Inyección de Ringer lactato. Inyección de Dextrosa 5%.
Sobredosificación.
Síntomas y signos: Somnolencia, agitación, agresión, disminución del nivel de conciencia, depresión respiratoria y coma fueron observados con la sobredosis por Levetiracetam. Tratamiento de la sobredosificación: En la sobredosis aguda está indicado el vaciado del contenido del estómago por lavado gástrico o por inducción de la emesis. No hay un antídoto específico para levetiracetam. El tratamiento de la sobredosificación será sintomático y puede incluir hemodiálisis. La eficacia de la eliminación por diálisis es del 60% para el levetiracetam y del 74% para el metabolito primario.
Presentación.
CEUMID 500: Envase conteniendo 30 comprimidos recubiertos. CEUMID 1000: Envase conteniendo 30 comprimidos recubiertos. CEUMID solución oral: Frasco conteniendo 150 mL y copa dosificadora. CEUMID IV: Envase conteniendo 10 viales de 5 ml de concentrado para solución para perfusión cada uno.


 Cambiar de país
Cambiar de país