ACTEMRA®
ROCHE
El tocilizumab es un anticuerpo monoclonal recombinante humanizado de la subclase IgG1 de las inmunoglobulinas (Ig), dirigido contra el receptor de la interleucina 6 (IL-6) humana. Clasificación: Inhibidores de la interleucina. Código ATC: L04AC07.
Composición.
Principio activo: tocilizumab. El tocilizumab es un líquido entre límpido y opalescente y entre incoloro y amarillo pálido, presentado en viales para un solo uso apirógenos y sin conservantes. El tocilizumab se presenta en viales de 10 ml y 20 ml con 4 ml, 10 ml o 20 ml de tocilizumab (20 mg/ml). Excipientes: polisorbato 80, sacarosa, fosfato disódico dodecahidrato, dihidrogenofosfato de sodio dihidrato y agua para inyectables. Forma farmacéutica: Concentrado para solución para infusión. Vía de administración: Infusión intravenosa (i.v). Declaración de esterilidad / radiactividad: Producto estéril.
Farmacología.
Propiedades farmacodinámicas: En los estudios clínicos con el tocilizumab se observaron descensos rápidos de la proteína C-reactiva (PCR), la velocidad de sedimentación globular (VSG) y la proteína amiloide A sérica. Se observó un aumento de la hemoglobinemia debido a que el tocilizumab reduce los efectos de la IL-6 sobre la producción de hepcidina y se incrementa así la disponibilidad del hierro. En sujetos sanos que recibieron dosis de tocilizumab entre 2 y 28 mg/kg, el recuento absoluto de neutrófilos descendió al nivel más bajo al cabo de 3-5 días de la administración. Después, la cifra de neutrófilos retornó hacia el recuento inicial (basal) en función de la dosis. En los pacientes con AR fue similar el patrón del recuento absoluto de neutrófilos tras la administración de tocilizumab (ver Pruebas de laboratorio). Mecanismo de acción: El tocilizumab es un anticuerpo monoclonal recombinante humanizado de la subclase IgG1 de las inmunoglobulinas (Ig), dirigido contra el receptor de la interleucina 6 (IL-6) humana. El tocilizumab se une específicamente tanto a los receptores solubles de la IL-6 (RIL-6s) como a los receptores membranarios (RIL-6m), y se ha constatado que inhibe la transmisión de señales mediada por RIL-6s y RIL-6m. La IL-6 es una citocina multifuncional, producida por diversos tipos celulares que intervienen en funciones paracrinas locales y en la regulación de procesos sistémicos fisiológicos y patológicos como la inducción de la secreción de inmunoglobulinas, la activación de linfocitos T, la inducción de proteínas hepáticas de fase aguda y la estimulación de la hematopoyesis. Se ha implicado a la IL-6 en la patogenia de enfermedades, entre ellas afecciones inflamatorias, osteoporosis y neoplasias. Es posible que el tocilizumab afecte a las defensas del huésped contra infecciones y neoplasias. No se sabe qué función desempeña la inhibición del receptor de la IL-6 en el desarrollo de neoplasias. Ensayos clínicos / Eficacia: Artritis reumatoide: Se ha evaluado la eficacia del tocilizumab en el alivio de los signos y síntomas de la AR en cinco estudios aleatorizados, multicéntricos y con doble enmascaramiento. En los estudios I-V se exigía que los pacientes tuvieran al menos 18 años, AR activa diagnosticada según los criterios del Colegio Estadounidense de Reumatología (ACR) y al menos 8 articulaciones dolorosas al tacto y 6 articulaciones tumefactas en la evaluación inicial. Se administró tocilizumab por vía intravenosa cada 4 semanas como monoterapia (estudio I) y en combinación con MTX (estudios II, III, V) o con otros FAMEs (estudio IV). En el estudio I se evaluó a 673 pacientes que no habían sido tratados con MTX en los 6 meses anteriores a la aleatorización y no habían suspendido anteriormente un tratamiento con MTX por efectos tóxicos clínicamente importantes o falta de respuesta. La mayoría de los pacientes (67%) no habían recibido nunca MTX. Se administraron dosis de 8 mg/kg de tocilizumab cada cuatro semanas como monoterapia. El grupo comparativo recibió MTX una vez por semana (dosis ajustada desde 7,5 mg hasta un máximo de 20 mg una vez por semana, a lo largo de 8 semanas). La variable principal de valoración era el porcentaje de pacientes con una respuesta ACR 20 en la semana 24. En el estudio II, un estudio de 2 años en marcha con análisis intermedio previsto en las semanas 24 y 52, se evaluó a 1.196 pacientes con una respuesta clínica insuficiente al MTX. Se administraron dosis de 4 u 8 mg/kg de tocilizumab o placebo cada cuatro semanas como tratamiento enmascarado durante 52 semanas, en combinación con MTX estable (10-25 mg por semana). La variable principal de valoración en la semana 24 era el porcentaje de pacientes con una respuesta ACR 20. En la semana 52, las variables coprincipales de valoración eran la prevención del daño articular y una mejora en la función física. En el estudio III se evaluó a 623 pacientes con una respuesta clínica insuficiente al MTX. Se administraron dosis de 4 u 8 mg/kg de tocilizumab o placebo cada cuatro semanas, en combinación con MTX estable (10-25 mg por semana). En el estudio IV se evaluó a 1.220 pacientes que no respondían suficientemente a sus tratamientos reumatológicos del momento, incluidos uno o más FAMEs. Se administraron dosis de 8 mg/kg de tocilizumab o placebo cada cuatro semanas, en combinación con el FAME estable. En el estudio V se evaluó a 499 pacientes con una respuesta clínica insuficiente o intolerancia a uno o más tratamientos anti-FNT. El fármaco anti-FNT se retiró antes de la aleatorización. Se administraron dosis de 4 u 8 mg/kg de tocilizumab o placebo cada cuatro semanas, en combinación con MTX estable (10-25 mg por semana). La variable principal de valoración en los estudios III-V era el porcentaje de pacientes con una respuesta ACR 20 en la semana 24. En la Tabla 2 se muestra el porcentaje de pacientes que lograron una respuesta ACR 20, 50 o 70 en los estudios I-V.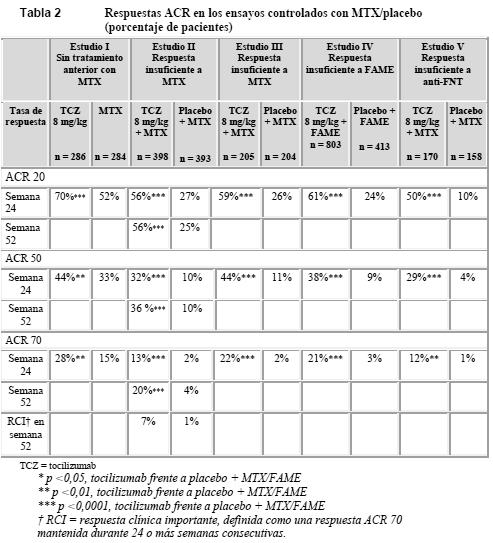
En todos los estudios, los pacientes tratados con 8 mg/kg de tocilizumab tuvieron tasas de respuesta ACR 20, 50 o 70 significativamente más elevadas a los 6 meses que los controles. El efecto terapéutico fue similar independientemente de factores como factor reumatoide, edad, sexo, raza, número de tratamientos anteriores o estado clínico de los pacientes. La rapidez del efecto fue grande (presente ya desde la segunda semana) y la magnitud de la respuesta siguió aumentando a lo largo del tratamiento. Se observaron respuestas duraderas y continuas durante más de 3 años en los estudios de extensión no enmascarados en marcha de los estudios I-V. En comparación con los pacientes que recibieron placebo + MTX/FAME, los tratados con 8 mg/kg de tocilizumab alcanzaron en todos los estudios mejoras significativas de todos los componentes individuales de la respuesta ACR (número de articulaciones dolorosas al tacto y tumefactas, evaluación global por el paciente y por el médico, puntuaciones del índice de incapacidad [HAQ], evaluación del dolor y PCR). El descenso del índice de actividad de la enfermedad (DAS 28) fue significativamente mayor en los pacientes tratados con 8 mg/kg de tocilizumab que en los que recibieron placebo + FAME. El número de pacientes que alcanzaron una respuesta EULAR entre buena y moderada fue significativamente mayor entre los tratados con tocilizumab que entre los que recibieron placebo + FAME (Tabla 3).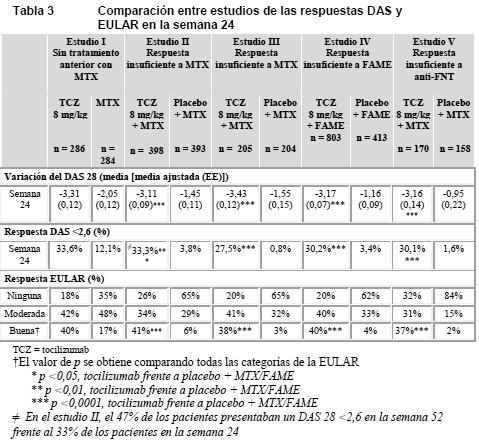
Respuesta clínica mayor: Al cabo de 2 años de tratamiento con tocilizumab/MTX, el 14% de los pacientes presentaban una respuesta clínica importante (mantenimiento de una respuesta ACR 70 durante 24 o más semanas). Respuesta radiográfica: En el estudio II, realizado en pacientes que no habían respondido adecuadamente al MTX, el daño estructural articular se evaluó radiográficamente y se expresó como variación del índice total de Sharp modificado y sus componentes, la puntuación de la erosión y la puntuación del estrechamiento del espacio articular. La inhibición del daño estructural articular se manifestó como progresión radiográfica significativamente menor en los pacientes tratados con tocilizumab que en los de control. En la extensión sin enmascaramiento del estudio II, la inhibición de la progresión de la lesión articular estructural en los pacientes tratados con tocilizumab/MTX se mantuvo en el segundo año de tratamiento.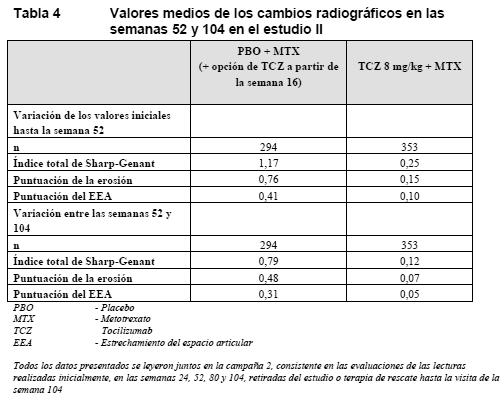
Tras 1 año de tratamiento con tocilizumab/MTX, el 83% de los pacientes no mostraban progresión alguna de la lesión articular estructural, definida como una variación de la puntuación total de cero o menor en la escala de Sharp, en comparación con el 67% de los pacientes que recibieron placebo/MTX. Este resultado se mantuvo constante tras 2 años de tratamiento (83%). El 93% de los pacientes no mostraron progresión alguna entre las semanas 52 y 104. Parámetros de la calidad de vida: Se observaron mejoras clínicamente significativas del índice de incapacidad (HAQ-DI, Health Assessment Questionnaire Disability Index) y de fatiga (FACIT-F, Functional Assessment of Chronic Illness Therapy Fatigue), así como mejoras de los componentes del cuestionario SF-36 (Short Form 36) correspondientes a la salud física (PCS, Physical Component Summary) y a la salud mental (MCS, Mental Component Summary), en los pacientes tratados con 8 mg/kg de tocilizumab (en monoterapia o en combinación con FAMEs), en comparación con los pacientes tratados con MTX/FAME (Tabla 5). En la semana 24, la proporción de pacientes con una mejora clínicamente relevante del HAQ-DI (definida como descenso de la puntuación total individual > 0,25) era significativamente mayor en los pacientes tratados con 8 mg/kg de tocilizumab que entre los tratados con placebo + MTX/FAME en todos los estudios. Durante el periodo sin enmascaramiento del estudio II, la mejora de la función física se mantuvo hasta dos años.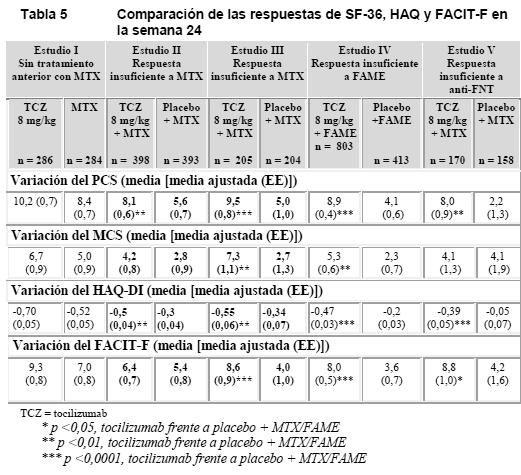
En el estudio II, las variaciones de los valores de PCS, MCS y FACIT-F en la semana 52 fueron de 10,1***, 5,4 y 8,4**, respectivamente, en el grupo de TCZ 8 mg/kg + MTX frente a 5,6, 3,8 y 5,5, respectivamente, en el grupo de placebo + MTX. En la semana 52, la media de la variación del HAQ-DI fue de -0,58 en el grupo de TCZ 8 mg/kg + MTX y de -0,39 en el grupo de placebo + MTX. La media de la variación del HAQ-DI se mantuvo en la semana 104 en el grupo de TCZ 8 mg/kg + MTX (-0,61). Evaluaciones de laboratorio: El tratamiento con 8 mg/kg de tocilizumab en combinación con FAME/MTX o en monoterapia logró una mejoría muy significativa de la hemoglobinemia en comparación con placebo + MTX/FAME (p < 0,0001) en la semana 24. La mejoría más notable se observó en pacientes con anemia crónica asociada a la AR; los valores medios de la hemoglobinemia habían aumentado en la semana 2 y se mantuvieron dentro del intervalo normal hasta la semana 24. Tras la administración de tocilizumab se produjo rápidamente un acusado descenso de las concentraciones medias de reactantes de fase aguda, PCR, VSG y proteína amiloide A sérica. En consonancia con el efecto sobre los reactantes de fase aguda, el tratamiento con tocilizumab se acompañó de una reducción de la cifra de plaquetas dentro de los límites normales. Monoterapia: Tocilizumab versus adalimumab: El ensayo WA19924, evaluó a 326 pacientes con AR que eran intolerantes a MTX o donde el tratamiento continuado con MTX se consideraba inapropiado (incluyendo respondedores inadecuados a MTX). Los pacientes del grupo de tocilizumab recibieron una infusión intravenosa (IV) de tocilizumab (8 mg/kg) cada 4 semanas (q4w) y una inyección subcutánea (SC) de placebo cada 2 semanas (q2w). Los pacientes del grupo de adalimumab recibieron una inyección SC de adalimumab (40 mg) cada 2 semanas (q2w) más una infusión IV de placebo cada 4 semanas (q4w). Se observó un efecto de tratamiento superior, estadísticamente significativo de tocilizumab sobre adalimumab, en el control de la actividad de la enfermedad, desde el valor basal a la semana 24, para la variable primaria cambio en DAS28 y para todas las variables secundarias (Tabla 6).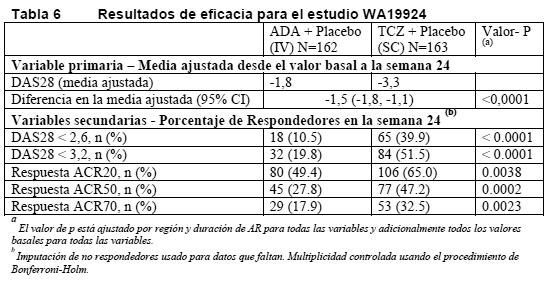
Artritis idiopática juvenil poliarticular: La eficacia del tocilizumab se evaluó en un estudio de tres partes que comprendía una de extensión no enmascarada en niños con AIJp. La parte I consistió en un periodo de preinclusión de tratamiento activo con tocilizumab (n = 188), a la que siguió una parte II, un periodo de retirada aleatorizado, con doble enmascaramiento y controlado con placebo, de 24 semanas (población ITT: n = 163), seguida de la parte III, un periodo no enmascarado de 64 semanas. Los pacientes seleccionables con un peso ≥30 kg recibieron 4 dosis de tocilizumab de 8 mg/kg. A los pacientes con < 30 kg de peso se los asignó aleatorizadamente 1:1 para recibir 8 mg/kg o 10 mg/kg de tocilizumab cada 4 semanas hasta completar 4 dosis. Los pacientes que finalizaron la parte I del estudio y alcanzaron una respuesta ACR 30 en AIJ en la semana 16 en relación con el valor inicial pasaron al periodo de retirada enmascarado (parte II) del estudio. En la parte II se aleatorizó a los pacientes 1:1 para recibir tocilizumab (igual dosis que en la parte I) o placebo y se los estratificó en función del uso concomitante de metotrexato o corticosteroides. Cada paciente continuó en la parte II del estudio hasta la semana 40 o hasta que satisfacía el criterio de exacerbación de ACR 30 en AIJ (en relación con la semana 16) y estaba cualificado para la terapia de rescate. La variable principal de valoración era la proporción de pacientes con exacerbación de ACR 30 en AIJ en la semana 40 en la relación con la semana 16. El 48,1% de los pacientes (39 de 81) que recibieron placebo experimentaron una exacerbación frente al 25,6% (21 de 82) de los tratados con TCZ. Estas proporciones diferían en grado estadísticamente significativo (p = 0,0024). Al final de la parte I, las respuestas ACR 30, 50, 70 o 90 en AIJ eran del 89,4%, 83,0%, 62,2% y 26,1%, respectivamente. En la tabla siguiente se muestran los porcentajes de pacientes con una respuestas ACR 30, 50 o 70 en AIJ en la semana 40 en relación con los valores iniciales durante la fase de retirada (parte II).
Artritis idiopática juvenil sistémica: La eficacia del tocilizumab en el tratamiento de la AIJs se evaluó en un estudio de 12 semanas aleatorizado, con doble enmascaramiento, controlado con placebo y de grupos paralelos. Se aleatorizó (TCZ:placebo = 2:1) a los pacientes (tratados con o sin MTX) en dos grupos: uno de 75 pacientes que recibieron infusiones de tocilizumab cada dos semanas, bien 8 mg/kg en los pacientes con un peso ≥30 kg o 12 mg/kg en los pacientes con un peso < 30 kg, y otro de 37 pacientes que recibieron infusiones de placebo cada dos semanas. Se permitió la disminución progresiva de corticosteroides a partir de la semana seis en los pacientes con una respuesta ACR 70 en AIJ. Al cabo de las 12 semanas o en el momento de la terapia de rescate, a causa de empeoramiento de la enfermedad, los pacientes recibieron tratamiento en la fase de extensión no enmascarada con dosis adecuadas para su peso. La variable principal de valoración era la proporción de pacientes con una mejora de al menos el 30% en el conjunto de datos básicos ACR en AIJ (respuesta ACR 30 en AIJ) en la semana 12 y ausencia de fiebre (ningún registro ≥37,5°C en los 7 días previos). El 85% de los pacientes (64 de 75) tratados con TCZ y el 24,3% (9 de 37) de los que recibieron placebo alcanzaron esta variable de valoración. La diferencia estadística de estas proporciones era altamente significativa (p < 0,0001). En la tabla siguiente se muestra el porcentaje de pacientes con una respuesta ACR 30, 50, 70 o 90 en AIJ. Las respuestas se mantienen en la fase de extensión no enmascarada.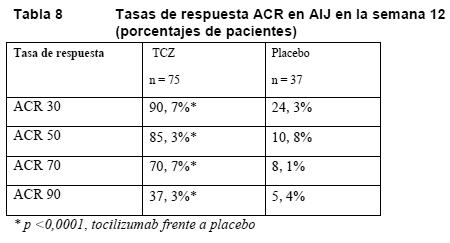
Características sistémicas: Entre los pacientes tratados con tocilizumab, el 85% de los que presentaban fiebre inicialmente a causa de la AIJs estaban apiréticos (ningún registro ≥37,5°C en los 14 días previos) en la semana 12 frente al 21% de los pacientes del grupo de placebo (p < 0,0001), y el 64% de los tratados con tocilizumab con erupción típica de la AIJs inicialmente no presentaban ninguna erupción en la semana 12 frente al 11% de los pacientes del grupo de placebo (p = 0,0008). La disminución del dolor en la semana 12 en los pacientes tratados con tocilizumab era altamente significativa estadísticamente en comparación con los que recibieron placebo. La variación media ajustada del dolor después de 12 semanas de tratamiento con tocilizumab alcanzó un descenso de 41 puntos en una escala visual analógica de 0-100 puntos frente a un descenso de 1 punto en los pacientes del grupo de placebo (p < 0,0001). Las respuestas relativas a las características sistémicas se mantenían en la fase de extensión no enmascarada en marcha. Disminución progresiva de corticosteroides: De los 31 pacientes del grupo de placebo y los 70 del grupo de tocilizumab que recibían corticosteroides orales inicialmente, 8 del grupo de placebo y 48 del grupo de tocilizumab alcanzaron una respuesta ACR 70 en AIJ en la semana 6 u 8 que permitió la reducción progresiva de los corticosteroides. Diecisiete (24%) pacientes del grupo de tocilizumab frente a 1 (3%) del grupo de placebo pudieron reducir la dosis de corticosteroides en al menos el 20% sin sufrir después una exacerbación de ACR 30 en AIJ o síntomas sistémicos hasta la semana 12 (p = 0,028). La reducción de los corticosteroides continuó, de modo que en la semana 44 había 44 pacientes que no recibían ningún corticosteroide oral, pero mantenían la respuesta ACR. Calidad de vida: En la semana 12, la proporción de pacientes con una mejora clínicamente importante aunque mínima del HAQ-DI (definida como descenso de la puntuación total individual ≥0,13) era significativamente mayor en los pacientes tratados con tocilizumab que en los del grupo de placebo (77% frente al 19%, p < 0,0001). Las respuestas se mantenían en la fase de extensión no enmascarada en marcha. Parámetros de laboratorio: Cincuenta de 76 (67%) los pacientes tratados con tocilizumab presentaban inicialmente un valor de hemoglobina < LIN (límite inferior de la normalidad). Cuarenta (80%) de estos pacientes con cifras de hemoglobina reducidas experimentaron un aumento de las mismas hasta valores normales en la semana 12 frente a sólo 2 de 29 (7%) pacientes del grupo de placebo con valores iniciales de hemoglobina < LIN (p < 0,0001). Cuarenta y cuatro (88%) pacientes tratados con tocilizumab con valores iniciales de hemoglobina reducidos experimentaron un aumento de los mismos ≥10 g/l en la semana 6 frente a 1 (3%) paciente del grupo de placebo (p < 0,0001). La proporción de pacientes del grupo de tocilizumab con trombocitosis inicialmente que presentaban un recuento normal de plaquetas en la semana 12 era significativamente mayor que en el grupo de placebo (90% frente al 4%, p < 0,0001). Tras la administración de tocilizumab se produjo rápidamente un acusado descenso de las concentraciones medias de reactantes de fase aguda, PCR, VSG y proteína amiloide A sérica. Propiedades farmacocinéticas: Artritis reumatoide: La farmacocinética del tocilizumab se determinó mediante un análisis de farmacocinética poblacional utilizando una base de datos integrada por 1.793 pacientes con AR tratados con una infusión de 4 y 8 mg/kg de una hora de duración, cada 4 semanas, durante 24 semanas. Los parámetros farmacocinéticos del tocilizumab no cambiaban con el tiempo. Se observó un aumento no lineal, mayor que el aumento de la dosis, del área bajo la curva de concentraciones plasmáticas (ABC) y de la concentración mínima (Cmín) con las dosis de 4 y 8 mg/kg cada 4 semanas. La concentración máxima (Cmáx) aumentó de forma lineal en relación con la dosis. En el estado de equilibrio estacionario, el ABC y la Cmín previstas eran, respectivamente, 2,7 y 6,5 veces más altas con la dosis de 8 mg/kg que con la de 4 mg/kg. Los parámetros siguientes son válidos para una dosis de tocilizumab de 8 mg/kg administrada cada 4 semanas. En equilibrio estacionario, la media (± DE) prevista del ABC, la Cmín y la Cmáx de tocilizumab era de 35.000 ± 15.500 mcg•h /ml, 9,74 ± 10,5 mcg/ml y 183 ± 85,6 mcg/ml, respectivamente. Los cocientes de acumulación del ABC y la Cmáx eran pequeños: 1,22 y 1,06, respectivamente. El cociente de acumulación era mayor para la Cmín (2,35), lo que cabía esperar dada la contribución del aclaramiento no lineal con concentraciones más bajas. Se alcanzó el equilibrio estacionario tras la primera administración en el caso de la Cmáx y tras 8 y 20 semanas en el caso del ABC y la Cmín, respectivamente. El ABC, la Cmín y la Cmáx de tocilizumab se elevaban con el aumento del peso corporal. Con un peso ≥100 kg, la media (± DE) prevista del ABC, la Cmín y la Cmáx en equilibrio estacionario de tocilizumab era de 55.500 ± 14.100 mcg•h/ml, 19,0 ± 12,0 mcg/ml y 269 ± 57 mg/ml, respectivamente, valores que son más altos que los valores medios de exposición en los pacientes. Por tanto, no se recomiendan dosis superiores a 800 mg por infusión en los pacientes con un peso ≥100 kg (ver Dosificación). Los parámetros siguientes son válidos para una dosis de tocilizumab de 4 mg/kg administrada cada 4 semanas. En equilibrio estacionario, la media (± DE) prevista del ABC, la Cmín y la Cmáx de tocilizumab era de 13.000 ± 5.800 mcg·h/ml, 1,49 ± 2,13 mcg/ml y 88,3 ± 41,4 mcg/ml, respectivamente. Los cocientes de acumulación del ABC y la Cmáx eran pequeños: 1,11 y 1,02, respectivamente. El cociente de acumulación era mayor para la Cmín (1,96). El equilibrio estacionario se alcanzó tras la primera administración en el caso de la Cmáx y el ABC y al cabo de 16 semanas en el caso de la Cmín. Artritis idiopática juvenil poliarticular: La farmacocinética del tocilizumab se determinó mediante un análisis de farmacocinética poblacional utilizando una base de datos integrada por 188 pacientes con AIJp. Los parámetros siguientes son válidos para una dosis de tocilizumab de 8 mg/kg (peso corporal ≥30 kg) administrada cada 4 semanas. La media (± DE) prevista del ABC4sem, la Cmáx y la Cmín de tocilizumab era de 29.500 ± 8.660 h•mcg/ml, 182 ± 37 mcg/ml y 7,49 ± 8,2 mcg/ml, respectivamente. Los parámetros siguientes son válidos para una dosis de tocilizumab de 10 mg/kg (peso corporal < 30 kg) administrada cada 4 semanas. La media (± DE) prevista del ABC4sem, la Cmáx y la Cmín de tocilizumab era de 23.200 ± 6.100 mcg•h/ml, 175 ± 32 mcg/ml y 2,35 ± 3,59 mcg/ml, respectivamente. El cociente de acumulación era de 1,05 y 1,16 para el ABC4sem, y de 1,43 y 2,22 para la Cmín con dosis de 10 mg/kg (peso corporal < 30 kg) y 8 mg/kg (peso corporal ≥30 kg), respectivamente. No se observó acumulación en la Cmáx. Artritis idiopática juvenil sistémica: La farmacocinética del tocilizumab se determinó mediante un análisis de farmacocinética poblacional utilizando una base de datos integrada por 75 pacientes con AIJs tratados con dosis de 8 mg/kg (peso corporal ≥30 kg) o 12 mg/kg (peso corporal < 30 kg) cada 2 semanas. La media (± DE) prevista del ABC2sem, la Cmáx y la Cmín de tocilizumab era de 32.200 ± 9.960 h•mg/ml, 245 ± 57,2 mg/ml y 57,5 ± 23,3 mg/ml, respectivamente. El cociente de acumulación era de 3,2 ± 1,3 para la Cmín (semana 12/semana 2). La Cmín de tocilizumab se estabilizó después de la semana 12. Los parámetros de la exposición media prevista al tocilizumab eran similares en uno y otro grupo de peso. Distribución: Tras la administración i.v., el tocilizumab es eliminado de la circulación sanguínea en un proceso bifásico. En los pacientes con AR, el volumen de distribución central era de 3,5 litros y el volumen de distribución periférico era de 2,9 litros, lo que suma un volumen de distribución en equilibrio de 6,4 litros. En los pacientes pediátricos con AIJp, el volumen de distribución central era de 1,98 litros y el volumen de distribución periférico era de 2,1 litros, lo que suma un volumen de distribución en equilibrio de 4,08 litros. En los pacientes pediátricos con AIJs, el volumen de distribución central era de 0,94 litros y el volumen de distribución periférico era de 1,60 litros, lo que suma un volumen de distribución en equilibrio de 2,54 litros. Eliminación: El aclaramiento total del tocilizumab dependía de la concentración y es la suma del aclaramiento lineal y del no lineal. Se calculó el aclaramiento lineal como un parámetro del análisis de farmacocinética poblacional y era de 12,5 ml/h en los pacientes con AR, 5,8 ml/h en los pacientes pediátricos con AIJp y de 7,1 ml/h en los pacientes pediátricos con AIJs. El aclaramiento no lineal dependiente de la concentración desempeña un papel importante cuando las concentraciones de tocilizumab son bajas. Una vez saturada la vía del aclaramiento no lineal, con concentraciones de tocilizumab más altas el aclaramiento viene determinado principalmente por el aclaramiento lineal. La semivida de eliminación (t1/2) del tocilizumab depende de la concentración en la AR; la t1/2 aparente dependiente de la concentración es de hasta 11 días con la dosis de 4 mg/kg y de 13 días con la de 8 mg/kg cada 4 semanas en equilibrio estacionario. La t1/2 del tocilizumab en pacientes pediátricos con AIJp es de hasta 16 días en ambos grupos de peso (8 mg/kg con un peso ≥30 kg o 10 mg/kg con un peso < 30 kg) en equilibrio estacionario. La t1/2 del tocilizumab en pacientes pediátricos con AIJs es de hasta 23 días en ambos grupos de peso (8 mg/kg con un peso ≥30 kg o 12 mg/kg con un peso < 30 kg) en la semana 12. Farmacocinética en poblaciones especiales: Insuficiencia hepática: No se ha realizado ningún estudio formal sobre el efecto de la insuficiencia hepática en la farmacocinética del tocilizumab. Insuficiencia renal: No se ha realizado ningún estudio formal sobre el efecto de la insuficiencia renal en la farmacocinética del tocilizumab. La mayoría de los pacientes del análisis de farmacocinética poblacional tenían una función renal normal o una insuficiencia renal leve. La insuficiencia renal leve (aclaramiento de creatinina basado en la fórmula de Cockcroft-Gault < 80 ml/min y ≥50 ml/min) no modificaba la farmacocinética del tocilizumab. No es necesario ajustar la dosis en los pacientes con insuficiencia renal leve o moderada. Otras poblaciones especiales: Los análisis de farmacocinética poblacional en pacientes adultos con AR han puesto de manifiesto que la edad, el sexo y la raza no influyen en la farmacocinética del tocilizumab. No es necesario ajustar la dosis por estos factores demográficos. Datos preclínicos sobre seguridad: Carcinogenicidad: No se han llevado a cabo estudios sobre la carcinogenicidad del tocilizumab. Los datos preclínicos disponibles mostraban la contribución de la citocina pleiotrópica IL-6 a la evolución maligna y la resistencia a la apoptosis de diversos tipos de cáncer. Estos datos no indican que exista un riesgo significativo de aparición o progresión de una neoplasia durante el tratamiento con tocilizumab. En consonancia con ello, en un estudio de toxicidad crónica con dosis múltiples, de 6 meses de duración y realizado en macacos de Java no se han observado lesiones proliferantes, ni se han descrito tampoco en ratones con el gen de la IL-6 desactivado y deficiencia crónica de IL-6. Mutagenicidad: Todos los estudios convencionales de genotoxicidad del tocilizumab en células procariotas y eucariotas arrojaron resultados negativos. Trastornos de la fecundidad: Los datos preclínicos no denotan ningún efecto en la fecundidad del tratamiento con un análogo del tocilizumab. En un estudio de toxicidad crónica con dosis múltiples en macacos de Java no se observaron efectos sobre órganos endócrinos activos ni sobre los órganos del sistema reproductor, y tampoco quedó afectada la función reproductora en ratones de ambos sexos con deficiencia de IL-6. Teratogenicidad: Cuando se administró tocilizumab por vía intravenosa a hembras del macaco de Java al comienzo de la gestación, no se observaron efectos nocivos directos ni indirectos sobre el embarazo o el desarrollo embriofetal. Otros efectos: En un estudio de toxicidad embriofetal realizado en macacos de Java se observó un ligero incremento de abortos/muertes embriofetales con altas exposiciones sistémicas acumulativas ( > 100 veces la exposición humana) en el grupo que había recibido la dosis alta (50 mg/kg/día) en comparación con el grupo de placebo y otros grupos de dosis bajas. La tasa de abortos se hallaba dentro de los límites históricos para el macaco de Java en cautividad y los casos individuales de aborto o muerte embriofetal no presentaban ninguna relación sistemática con la dosis y la duración de la administración de tocilizumab. Aunque la IL-6 no parece ser una citocina esencial para el desarrollo fetal ni para el control inmunitario de la interfase maternofetal, no puede descartarse una relación entre estos datos y el tocilizumab. Se ha observado el paso de un análogo murino del tocilizumab a la leche de ratonas lactantes. El tratamiento con un análogo murino no fue tóxico en ratones jóvenes. Particularmente, no se deterioró el crecimiento óseo, la función inmunitaria ni la maduración sexual.
Indicaciones.
Artritis reumatoide: El tocilizumab está indicado para el tratamiento de la artritis reumatoide (AR) activa de grado entre moderado y grave en pacientes adultos que han tenido una respuesta inadecuada o intolerancia a un tratamiento previo con uno o más fármacos antirreumáticos modificadores de la enfermedad (FAMEs) o con antagonistas del factor de necrosis tumoral (TNF). El tocilizumab puede utilizarse solo o combinado con metotrexato (MTX) u otros fármacos antirreumáticos modificadores de la enfermedad (FAMEs). Artritis idiopática juvenil poliarticular: El tocilizumab está indicado para el tratamiento de la artritis idiopática juvenil poliarticular (AIJp) en pacientes de dos o más años de edad. El tocilizumab puede utilizarse solo o en combinación con MTX. Artritis idiopática juvenil sistémica: El tocilizumab está indicado para el tratamiento de la artritis idiopática juvenil sistémica (AIJs) en pacientes de dos o más años de edad. El tocilizumab puede utilizarse solo o en combinación con MTX.
Dosificación.
Artritis reumatoide (AR): Instrucciones generales: La dosis recomendada de tocilizumab para pacientes adultos es de 8 mg/kg una vez cada cuatro semanas en infusión i.v. Puede utilizarse solo o combinado con metotrexato (MTX) u otros FAMEs. El tocilizumab debe diluirlo un profesional sanitario hasta 100 ml con una solución salina al 0,9% m/v estéril, aplicando una técnica aséptica (ver Instrucciones especiales de uso, manipulación y eliminación). Se recomienda que la infusión i.v. de tocilizumab tenga una duración de una hora. Para las personas con un peso superior a los 100 kg no se recomiendan dosis superiores a 800 mg por infusión (ver Propiedades farmacocinéticas). Recomendaciones para modificar la dosis en la AR: (ver Pruebas de laboratorio).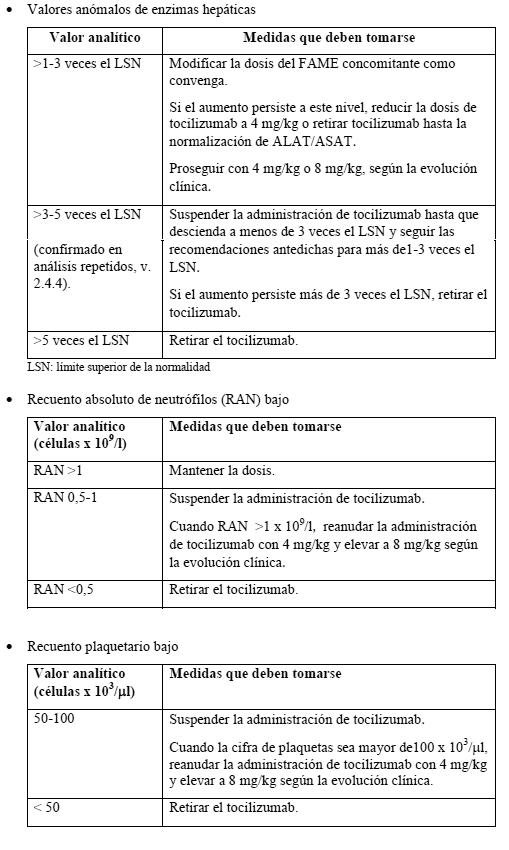
Artritis idiopática juvenil poliarticular: Dosis recomendada de tocilizumab en la AIJp: 10 mg/kg en los pacientes con un peso < 30 kg. 8 mg/kg en los pacientes con un peso ≥30 kg. En infusión i.v., cada cuatro semanas. Todo cambio de la dosis debe basarse exclusivamente en un cambio sostenido del peso del paciente a lo largo del tiempo. El tocilizumab puede utilizarse solo o en combinación con MTX. El tocilizumab debe diluirlo un profesional sanitario con una solución salina al 0,9% m/v estéril, aplicando una técnica aséptica (ver Instrucciones especiales de uso, manipulación y eliminación). Artritis idiopática juvenil sistémica: Dosis recomendada de tocilizumab en la AIJs: 12 mg/kg en los pacientes con un peso < 30 kg. 8 mg/kg en los pacientes con un peso ≥30 kg. Administrada una vez cada dos semanas, en infusión i.v. Todo cambio de la dosis debe basarse únicamente en un cambio sostenido del peso del paciente. El tocilizumab puede utilizarse solo o en combinación con metotrexato (MTX). El tocilizumab debe diluirlo un profesional sanitario con una solución salina al 0,9% m/v estéril, aplicando una técnica aséptica (ver Instrucciones especiales de uso, manipulación y eliminación). Se recomienda que la infusión i.v. de tocilizumab tenga una duración de una hora. Recomendaciones para modificar la dosis en la AIJp y la AIJs: En pacientes con AIJp o AIJs no se ha estudiado la reducción de la dosis de tocilizumab. En caso de alteraciones analíticas en pacientes con AIJp o AIJs, se recomienda interrumpir la administración de tocilizumab según lo expuesto para los pacientes con AR (ver Pruebas de laboratorio). Si se estima oportuno, puede modificarse o suspenderse la administración de MTX y/o de otros medicamentos concomitantes e interrumpirse la de tocilizumab hasta la evaluación de la situación clínica. En la AIJp y la AIJs, la decisión de retirar el tocilizumab por alteraciones analíticas debe basarse en el juicio médico del paciente afectado. Pautas posológicas especiales: Niños: La seguridad y la eficacia del tocilizumab se han estudiado únicamente en niños con AIJp o AIJs. Niños menores de dos años no han participado en los estudios. Ancianos: No es necesario ajustar la dosis en los pacientes de 65 o más años. Insuficiencia renal: No es necesario ajustar la dosis en pacientes con insuficiencia renal leve (ver Farmacocinética en poblaciones especiales). El tocilizumab no se ha estudiado en pacientes con insuficiencia renal moderada o grave. Insuficiencia hepática: No se han estudiado la seguridad y la eficacia del tocilizumab en pacientes con insuficiencia hepática (ver Advertencias).
Contraindicaciones.
Hipersensibilidad conocida al tocilizumab o a cualquiera de los excipientes.
Reacciones adversas.
Ensayos clínicos: Artritis reumatoide: La seguridad del tocilizumab se ha estudiado en 5 estudios de fase III controlados y con doble enmascaramiento (doble-ciego) y en sus períodos de extensión. La población global de control comprende a todos los pacientes de las fases con doble enmascaramiento de cada estudio central desde la aleatorización hasta el primer cambio de régimen terapéutico o hasta alcanzarse dos años. El periodo controlado en 4 estudios fue de 6 meses, y en 1, de hasta 2 años. En los estudios controlados con doble enmascaramiento, 774 pacientes recibieron 4 mg/kg de tocilizumab en combinación con MTX, 1.870 recibieron 8 mg/kg de tocilizumab en combinación con MTX/otro FAME y 288 recibieron 8 mg/kg de tocilizumab en monoterapia. La población global de exposición comprende a todos los pacientes que recibieron al menos una dosis de tocilizumab en el período controlado con doble enmascaramiento o en la fase de extensión no enmascarada de los estudios. De los 4.009 pacientes, 3.577 recibieron tratamiento durante un mínimo de 6 meses; 3.296, durante un mínimo de un año; 2.806, durante un mínimo de 2 años, y 1.222, durante 3 años. Las RA se enumeran según la importancia clínica para el paciente. Definición de la frecuencia de las RA: muy frecuente (≥1/10), frecuente (≥1/100 a < 1/10) y poco frecuente (≥1/1.000 a < 1/100).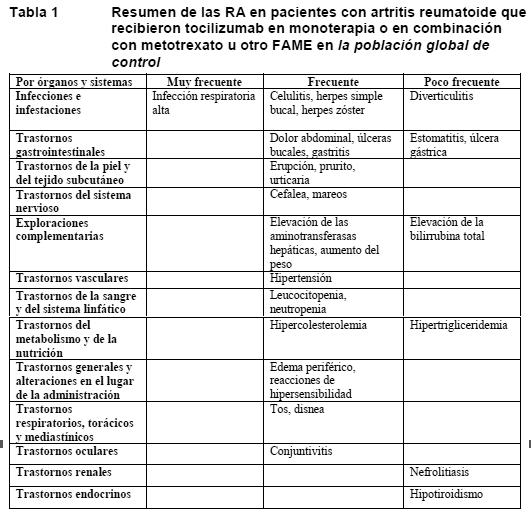
Infecciones: En los ensayos clínicos controlados de 6 meses, la tasa de todas las infecciones notificadas con el tratamiento de 8 mg/kg de tocilizumab + FAMEs fue de 127 episodios por 100 años-paciente frente a 112 episodios por 100 años-paciente en el grupo de placebo + FAMEs. En la población global de exposición, la tasa de infección con el tocilizumab fue de 108 episodios por 100 años-paciente de exposición. En los ensayos clínicos controlados de 6 meses, la tasa de infecciones graves (bacterianas, víricas y fúngicas) con 8 mg/kg de tocilizumab + FAMEs fue de 5,3 episodios por 100 años-paciente de exposición frente a 3,9 episodios por 100 años- paciente de exposición en el grupo que había recibido placebo + FAMEs. En el estudio de la monoterapia, la tasa de infecciones graves fue de 3,6 episodios por 100 años-paciente de exposición en el grupo de tocilizumab y de 1,5 episodios por 100 años-paciente de exposición en el del MTX. En la población global de exposición, la tasa de infecciones graves fue de 4,7 episodios por 100 años-paciente. Las infecciones graves -algunas con un desenlace fatal- notificadas consistieron en neumonía, celulitis, herpes zóster, gastroenteritis, diverticulitis, septicemia y artritis bacteriana. También se describieron infecciones oportunistas. Perforación gastrointestinal: Durante los estudios clínicos controlados de 6 meses, la tasa global de perforación gastrointestinal con tocilizumab fue de 0,26 episodios por 100 años-paciente. En la población global de exposición, la tasa global de perforación gastrointestinal fue de 0,28 episodios por 100 años-paciente. Las notificaciones de perforación gastrointestinal con tocilizumab se describieron principalmente como complicaciones de una diverticulitis e incluían peritonitis purulenta generalizada, perforación gastrointestinal baja, fístula y absceso. Reacciones a la infusión: En los estudios clínicos controlados de 6 meses se registraron acontecimientos adversos asociados con la infusión (determinados acontecimientos ocurridos durante la infusión o dentro de las 24 horas siguientes) en el 6,9% de los pacientes tratados con 8 mg/kg de tocilizumab + FAMEs y en el 5,1% de los que habían recibido placebo + FAMEs. Los acontecimientos notificados durante la infusión fueron fundamentalmente episodios de hipertensión; los notificados dentro de las 24 horas siguientes a la infusión consistieron en cefalea y reacciones cutáneas (erupción, urticaria). Estos acontecimientos no fueron limitantes del tratamiento. La tasa de anafilaxia (observada en un total de 6 de 3.778 pacientes) fue varias veces más alta en el grupo de 4 mg/kg que en el de 8 mg/kg. En 13 de los 3.778 pacientes (0,3%) tratados con tocilizumab en los ensayos clínicos controlados y no enmascarados se notificaron reacciones alérgicas clínicamente importantes asociadas con el tocilizumab que exigieron suspender el tratamiento. Estas reacciones se observaron generalmente durante las infusiones segunda a quinta de tocilizumab (ver Advertencias). Inmunogenicidad: En los ensayos clínicos controlados de 6 meses se determinó la presencia de anticuerpos antitocilizumab en 2.876 pacientes. Se detectaron anticuerpos en 46 de ellos (1,6%), de los que 5 sufrieron una reacción alérgica médicamente importante que condujo a la retirada de la medicación. Treinta pacientes (1,1%) desarrollaron anticuerpos neutralizantes. Monoterapia: tocilizumab versus adalimumab: En la semana 24 de un ensayo doble-ciego (monoterapia con tocilizumab IV 8 mg/kg cada 4 semanas (N=162) en comparación con adalimumab SC 40 mg cada 2 semanas (N=162)), el perfil clínico global de eventos adversos fue similar entre tocilizumab y adalimumab. La proporción de pacientes con