ALPHANATE
GRIFOLS-CHILE
Antihemofílico.
Composición.
Frasco-ampolla liofilizado: Principio activo: Factor VIII 250 U.I., 500 U.I., 1000 U.I., 1500 U.I./frasco-ampolla; Factor Von Willebrand/Cofactor de Ristocetina No inferior a 400 U.I. FVW:RCo /1000 U.I. FVIII:C. Excipientes: Albúmina 0,3 - 0,9 g/100 ml; Arginina 50 - 200 mmol/l; Histidina 10 - 40 mmol/l. Jeringa prellenada con disolvente: Agua Estéril para Inyección: 250 U.I., 500 U.I., 5 ml; 1000 U.I., 1500 U.I. 10 ml. Forma farmacéutica: Polvo liofilizado para solución inyectable con disolvente. Frasco-ampolla conteniendo polvo liofilizado blanco o amarillo pálido y jeringa con agua estéril para inyección (disolvente).
Farmacología.
Propiedades farmacodinámicas: Grupo Farmacoterapéutico: Antihemorrágicos: Factores de Coagulación de la Sangre: Código ATC B02BD06. El Factor Antihemofílico (Factor VIII) y el Factor de von Willebrand (FVW) son constituyentes del plasma normal y se requieren para la coagulación. La administración de Alphanate incrementa temporalmente el nivel en plasma de Factor VIII, y de este modo minimiza el peligro de hemorragia. El Factor VIII es un cofactor esencial en la activación del Factor X, que conduce a la formación de trombina y fibrina. El FVW promueve la agregación plaquetaria y la adhesión de plaquetas al endotelio vascular dañado; sirve también como proteína transportadora estabilizante para la proteína procoagulante Factor VIII. Propiedades farmacocinéticas: Hemofilia A: En un estudio cruzado abierto y aleatorizado se compararon las farmacocinéticas de Alphanate y del mismo producto pero sin tratamiento térmico. Los pacientes de hemofilia A ( < 3 U.I. factor VIII/dl) recibieron dosis adecuadas para alcanzar unos niveles máximos de factor VIII en plasma de alrededor de 100 unidades/dl. Se tomaron las muestras de sangre hasta las 28 horas tras la infusión. Para Alphanate la semivida fue de 12,2 horas y la recuperación media in vivo fue de 86,5%. En estos estudios la AUC media fue de 895 U.I. h/dL, el tiempo de residencia medio de 9,9 horas y el aclaramiento de 4,4 ml/h/kg. Dependiendo del grado de la hemorragia, lesión o daño tisular la semivida biológica puede ser inferior. Esto debe tenerse en cuenta para determinar la dosis. Enfermedad de von Willebrand (EVW): Se efectuó un estudio cruzado de farmacocinética en 14 sujetos con EVW sin hemorragias (1 de Tipo 1, 2 de Tipo 2A, y 11 de Tipo 3), comparando la farmacocinética de Alphanate SD/HT (A-SD/HT) con una formulación anterior, Alphanate SD (A-SD), tratado con solvente-detergente pero no con calor. Los sujetos recibieron aleatoriamente con un mínimo de siete días de diferencia, una sola dosis intravenosa de A-SD y de A-SD/HT, de 60 U.I. FVW:RCo/kg (75 U.I. FVW:RCo/kg en los sujetos menores de 18 años). Los parámetros farmacocinéticos fueron similares para los dos preparados e indicaron que ambos son bioquímicamente equivalentes. El análisis farmacocinético de A-SD/HT en los 14 sujetos reveló los siguientes resultados: los niveles plasmáticos medios de FVW:RCo subieron desde 0,17 UI/dl [media: 0,2 ± 0,08 UI/dl; intervalo: 0,1 a 0,5 UI/dl] hasta una línea de base de 3,43 U.I./dl [media: 3,5 ± 1,47 U.I./dl; intervalo: 1,5 a 5,9 U.I./dl] a los 15 minutos de la infusión; los niveles plasmáticos medios de FVIII:C subieron desde 0,08 U.I./dl [media: 0,2 ± 0,34 U.I./dl; intervalo: 0,0 a 1,2 U.I./dl] hasta 2,14 U.I./dl [media: 2,4 ± 0,72 U.I./dl; intervalo: 1,4 a 3,9 U.I./dl]. El tiempo medio de sangrado (TS) antes de la infusión era de 30 minutos (media: 28,8 ± 4,41 minutos; intervalo: 13,5 a 30 minutos), y se redujo a 10,38 minutos (media: 10,4 ± 3,20 minutos; intervalo: 6 a 16 minutos) al cabo de 1 hora tras la infusión. Después de la infusión de A-SD/HT, las semividas medias para FVW:RCo, FVIII:C y FVW:Ag fueron de 6,91 horas (media: 7,46 ± 3,20 horas, intervalo: 3,68 a 16,22 horas); 20,87 horas (media: 21,52 ± 7,21 horas; intervalo: 7,19 a 32,20 horas) y 12,66 horas (media: 13,03 ± 2,12 horas, intervalo: 10,34 a 17,45 horas) respectivamente. Los incrementos medios de las recuperaciones in vivo de FVW:RCo y FVIII:C fueron de 3,12 (U.I./dl)/(U.I./kg) [media: 3,29 ± 1,46 (U.I./dl)/(U.I./kg); intervalo: 1,3 a 5,7 (U.I./dl)/(U.I./kg)] para FVW:RCo y 1,94 (U.I./dl)/(U.I./kg) [media: 2,14 ± 0,58 (U.I./dl)/(U.I./kg); intervalo: 1,3 a 3,3 (U.I./dl)/(U.I./kg)] para FVIII:C. Después de la infusión tanto de A-SD como de A-SD/HT, se observó un incremento en el tamaño de los multímeros de FVW y persistió como mínimo 24 horas. El acortamiento de tiempo de sangrado fue transitorio, durando menos de 6 horas tras el tratamiento y no tuvo relación con la presencia de multímeros de FVW de tamaño grande o intermedio. Datos preclínicos sobre seguridad: Factor VIII de coagulación de plasma humano y factor de von Willebrand (principios activos de Alphanate) son constituyentes normales del plasma humano y actúan de igual forma que las correspondientes proteínas endógenas. Los ensayos de toxicidad de dosis única no son relevantes ya que dosis elevadas producen sobrecarga. Los ensayos en animales de toxicidad en dosis repetidas son impracticables debido a la interferencia de las proteínas heterólogas con los anticuerpos que se producen.
Indicaciones.
Hemofilia A o Deficiencia Adquirida de Factor VIII: Alphanate está indicado para el control y prevención del sangrado en pacientes con deficiencia de Factor VIII debido a hemofilia A o a una deficiencia adquirida de Factor VIII. Enfermedad de von Willebrand: Alphanate, está indicado para procedimientos quirúrgicos y/o invasivos en pacientes adultos y pediátricos con Enfermedad de von Willebrand en las que la desmopresina (DDAVP) o es inefectiva o está contraindicada. No existe suficiente evidencia procedente de ensayos clínicos en pacientes con Enfermedad de von Willebrand severa (Tipo 3) sometidos a cirugía mayor.
Dosificación.
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia. Posología: Después de la reconstitución con el disolvente proporcionado Alphanate debe ser administrado por vía intravenosa en las tres horas siguientes a la reconstitución para evitar el potencial efecto patológico de una contaminación bacteriana inadvertida ocurrida durante la reconstitución. Alphanate se administra por inyección (se recomiendan jeringas de plástico desechables). Administrar a temperatura ambiente, no refrigerar tras la reconstitución y desechar cualquier resto de producto no utilizado en un contenedor de seguridad apropiado. Hemofilia A: Los requerimientos de dosificación y la frecuencia de las dosis se calculan en base a una respuesta inicial esperada de un incremento del 2% del FVIII:C normal por cada U.I. de FVIII:C/kg peso corporal administrada. El incremento in vivo de Factor VIII en plasma puede, por tanto, ser estimado multiplicando la dosis de Factor Antihemofílico Humano (FAH)/kilogramo de peso corporal (U.I. FVIII:C /kg) por 2%. De este modo, una dosis administrada de FAH de 50 U.I./kg se esperará que incremente el nivel de Factor VIII circulante un 100 % del normal (100 U.I./dl). Las siguientes fórmulas y ejemplos ilustran estos principios:
En la Tabla 1 se presentan las dosis siguientes como recomendaciones generales. Debe incidirse en que la dosis de Alphanate requerida para la hemostasia debe ser individualizada en función de las necesidades del paciente, la severidad de la deficiencia, la severidad de la hemorragia, la presencia de inhibidores, y el nivel deseado de FVIII. Lo adecuado del tratamiento debe evaluarse por la situación y los efectos clínicos, y por tanto, la dosis puede variar con cada caso individual.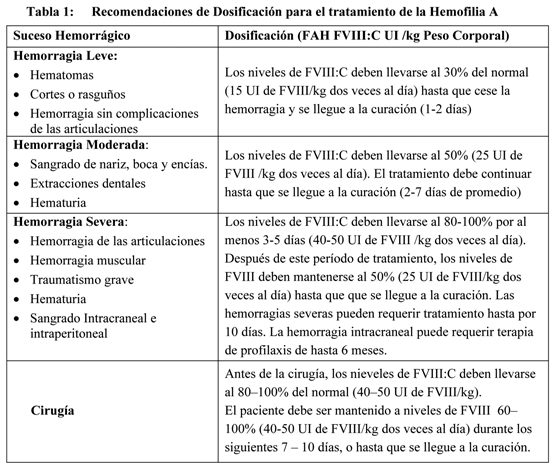
Los requerimientos de dosificación y la frecuencia de las dosis se calculan en base a una respuesta inicial esperada de un incremento del 2% de FVIII:C por U.I. de FVIII:C/kg peso corporal (por ej., 2% por U.I./kg) y una semivida media para FVIII:C de 12 horas. Si el estudio de la dosificación determina que un paciente en particular muestra una respuesta inferior a la esperada, la dosis debe ser ajustada adecuadamente. El fallo en alcanzar el nivel esperado de FVIII:C plasmático o en controlar el sangrado tras una dosis calculada adecuadamente, puede ser indicativo del desarrollo de un inhibidor (un anticuerpo para FVIII:C). Su presencia debe ser documentada y el nivel de inhibidor cuantificado mediante los procedimientos de laboratorio apropiados. En esos casos el tratamiento con FAH debe ser individualizado. Los niveles de factor VIII plasmático deben ser monitorizados periódicamente para evaluar la respuesta individual del paciente al régimen de dosificación. Enfermedad de Von Willebrand: La Tabla 2 proporciona las recomendaciones de dosificación para pacientes adultos y pediátricos con Enfermedad de von Willebrand. La actividad del Factor de von Willebrand:Cofactor de Ristocetina (FVW:RCo) se indica en unidades internacionales (U.I.) en la etiqueta del producto. La relación entre FVW:RCo y Factor VIII en Alphanate varía en cada lote, por lo que la dosificación debe ser reevaluada cada vez que se selecciona un lote diferente.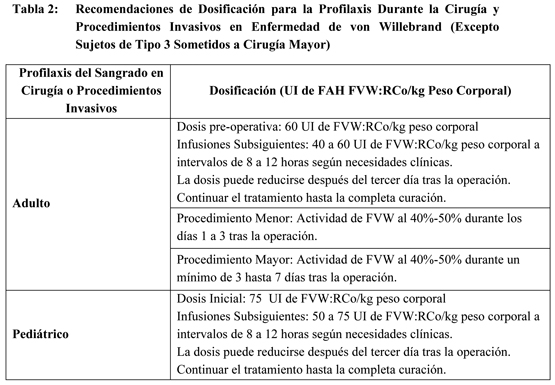
Contraindicaciones.
Hipersensibilidad al principio activo o a alguno de los excipientes (ver Advertencias.).
Reacciones adversas.
Las reacciones adversas serias observadas en pacientes que reciben Alphanate son anafilaxis o de hipersensibilidad (angioedema, sensación de ardor y picor en el lugar de la infusión, escalofríos, enrojecimiento, urticaria generalizada, cefalea, hipotensión, somnolencia, náuseas, inquietud, taquicardia, opresión torácica, hormigueo, vómitos, dificultad para respirar) y, en ciertos casos, estas reacciones han progresado hasta anafilaxia grave (incluyendo shock). Eventos tromboembólicos también han sido observados en pacientes que reciben Alphanate para la Enfermedad de Von Willebrand. Las reacciones adversas más frecuentes, reportadas con Alphanate en más del 5% de los pacientes son: dificultad respiratoria, prurito, rash, urticaria, edema facial, parestesia, dolor, fiebre, escalofríos, dolor articular y fatiga. Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizantes contra el factor VIII (inhibidores), lo que ocasiona una respuesta clínica insuficiente al tratamiento. En tales casos se recomienda contactar con un centro especializado en hemofilia. Actualmente no se dispone de suficiente información relacionada con el desarrollo de inhibidores en pacientes no previamente tratados con Alphanate. Ocasionalmente, pacientes con Enfermedad de von Willebrand, especialmente pacientes de tipo 3, pueden desarrollar anticuerpos neutralizantes contra el factor de von Willebrand, lo que ocasiona una respuesta clínica inadecuada al tratamiento. Estos inhibidores pueden producirse en asociación con reacciones anafilácticas. Por tanto, en los pacientes que experimenten una reacción anafiláctica debe evaluarse la presencia de inhibidores. En tales casos, se recomienda contactar con un centro especializado en hemofilia. Al usar este producto en pacientes con Enfermedad de von Willebrand existe riesgo de episodios trombóticos, especialmente en pacientes con factores de riesgo conocidos de tipo clínico o de laboratorio. En pacientes que reciben productos de factor VIII conteniendo factor de von Willebrand, mantener un excesivo nivel de FVIII:C puede incrementar el riesgo de sucesos trombóticos.
Advertencias.
Al igual que con cualquier producto proteico para administración intravenosa, es posible que se produzcan reacciones de hipersensibilidad de tipo alérgico. El producto contiene trazas de otras proteínas humanas además de factor VIII. Los pacientes deben ser informados acerca de los signos iniciales de las reacciones de hipersensibilidad, que incluyen erupciones cutáneas que pueden llegar a urticaria generalizada, opresión torácica, dificultad para respirar, hipotensión y anafilaxia. Si se producen reacciones de este tipo, se recomienda interrumpir la administración del preparado y contactar inmediatamente con el médico. En caso de shock, se seguirán las recomendaciones vigentes para tratamiento del shock. Las medidas empleadas para prevenir infecciones como consecuencia del uso de medicamentos elaborados a partir de sangre humana o de plasma incluyen la selección de los donantes de plasma, las pruebas en las donaciones individuales y en las mezclas de plasma, y la aplicación de etapas efectivas de eliminación/reducción víral. A pesar de estas medidas, cuando se administran productos preparados a partir de sangre humana o de plasma, la posibilidad de transmitir agentes infecciosos no puede ser totalmente excluida. Esto también puede aplicarse a virus emergentes u otro tipo de patógenos. Los procedimientos de inactivación/eliminación empleados se consideran efectivos para virus encapsulados como VIH, VHB y VHC. Estos procedimientos pueden tener un valor limitado para virus no encapsulados tales como VHA y Parvovirus B19. La infección por parvovirus B19 puede ser grave para una mujer embarazada (infección fetal) y para sujetos con inmunodeficiencia o con una producción aumentada de hematíes (ej. con anemia hemolítica). Se recomienda la vacunación apropiada (hepatitis A y B) para los pacientes que reciban de forma regular productos de factor VIII derivados de plasma humano. La formación de anticuerpos neutralizantes, o inhibidores, al factor VIII es una complicación conocida en los individuos con hemofilia A. Estos inhibidores son normalmente inmunoglobulinas IgG dirigidas contra la actividad procoagulante del factor VIII, que se cuantifica en unidades Bethesda (UB) por ml de plasma, a través del ensayo de Nijmegen modificado. El riesgo de desarrollar inhibidores está correlacionado con la exposición al factor VIII antihemofílico, estando el riesgo más alto en los 20 primeros días de exposición. Los inhibidores raramente se desarrollan tras los primeros 100 días. Los pacientes que reciben tratamiento con factor VIII de coagulación humano deben ser cuidadosamente monitorizados para observar si se desarrollan inhibidores mediante las observaciones clínicas y tests de laboratorio apropiados. Ver también Reacciones Adversas. Existen informes en la literatura que sugieren que los pacientes con Enfermedad de von Willebrand severa de tipo 3 ocasionalmente pueden desarrollar aloanticuerpos al factor de von Willebrand. Se recomienda que, cada vez que se administre Alphanate a un paciente, se deje constancia del nombre del medicamento y n° de lote administrado para establecer una relación entre el paciente y el lote del producto.
Interacciones.
No se han descrito interacciones de Alphanate con otros medicamentos. Embarazo y lactancia: No se han realizado estudios de reproducción en animales con Alphanate. Tampoco se conoce si Alphanate puede causar daño fetal si se administra a una mujer embarazada o si afecta la capacidad reproductiva. Alphanate debe administrarse a mujeres embarazadas solo si está claramente indicado. Efectos sobre la capacidad para conducir y utilizar máquinas: La influencia de Alphanate sobre la capacidad para conducir y utilizar máquinas es nula.
Incompatibilidades.
Alphanate no debe mezclarse con otros medicamentos. Unicamente debe utilizarse el equipo para inyección que se suministra para evitar un posible error en el tratamiento como consecuencia de la adsorción del Factor Von Willebrand/Cofactor de Ristocetina a la superficie interna de cualquier otro equipo para inyección.
Conservación.
Conservar entre 2°C y 8°C (en refrigerador). No congelar. El producto se puede conservar a temperatura ambiente, no superior a 30°C, por un periodo de hasta dos meses. Período de validez: El período de validez de Alphanate es de 2 años cuando se conserva entre 2°C y 8°C. La solución reconstituida debe ser utilizada inmediatamente o en el plazo de 3 horas. Precauciones especiales de eliminación y otras manipulaciones: No usar el producto después de la fecha de caducidad indicada en la etiqueta del frasco-ampolla. Antes de su uso, comprobar con atención el valor de la concentración del producto en la etiqueta. Utilizar técnicas asépticas durante la reconstitución y administración. El producto sobrante nunca debe ser guardado para su uso posterior, ni tampoco conservado en un refrigerador. Preparación de la Solución: 1. Atemperar el frasco-ampolla y la jeringa del disolvente sin sobrepasar los 30°C. 2. Acoplar el émbolo de plástico a la jeringa del disolvente. 3. Desprecintar el filtro. Separar el tapón del cono de la jeringa del disolvente y acoplarla al filtro. 4. Desprecintar el adaptador de frasco-ampolla y acoplarlo al conjunto filtro-jeringa. 5. Desprecintar el frasco-ampolla de concentrado, desinfectando el tapón con la toallita desinfectante suministrada. 6. Colocar el conjunto filtro/jeringa/adaptador sobre la parte superior del frasco-ampolla de concentrado y perforar el tapón con la aguja del adaptador. 7. Traspasar todo el Agua Estéril para Inyección dentro del frasco-ampolla de concentrado haciendo descender el émbolo de la jeringa. 8. Agitar suavemente el frasco-ampolla hasta que todo el concentrado esté disuelto. Como con otras soluciones parenterales, no usar la solución si no está adecuadamente disuelta o hay partículas visibles. 9. Separar brevemente el conjunto filtro/jeringa del resto para eliminar cualquier posible vacío. 10. Invertir el frasco-ampolla de concentrado y aspirar la solución a la jeringa a través del filtro. 11. Preparar la zona de inyección del paciente, separar la jeringa del resto. Inyectar la solución por vía intravenosa usando la aguja mariposa con cánula suministrada o una aguja estéril. Administrar lentamente a una velocidad no superior a los 10 ml/minuto. La administración rápida de un concentrado de Factor VIII puede provocar reacciones vasomotoras. Tras la reconstitución con el disolvente Agua Estéril para Inyección proporcionado, el producto debe ser usado inmediatamente. No reutilizar los kits de administración. Cualquier resíduo de producto no utilizado y el material de desecho debe ser eliminado de acuerdo con las regulaciones locales. La solución debe ser clara o ligeramente opalescente. No utilizar soluciones turbias o con depósitos. El producto reconstituido debe ser inspeccionado visualmente para ver la presencia de partículas y decoloración antes de su administración.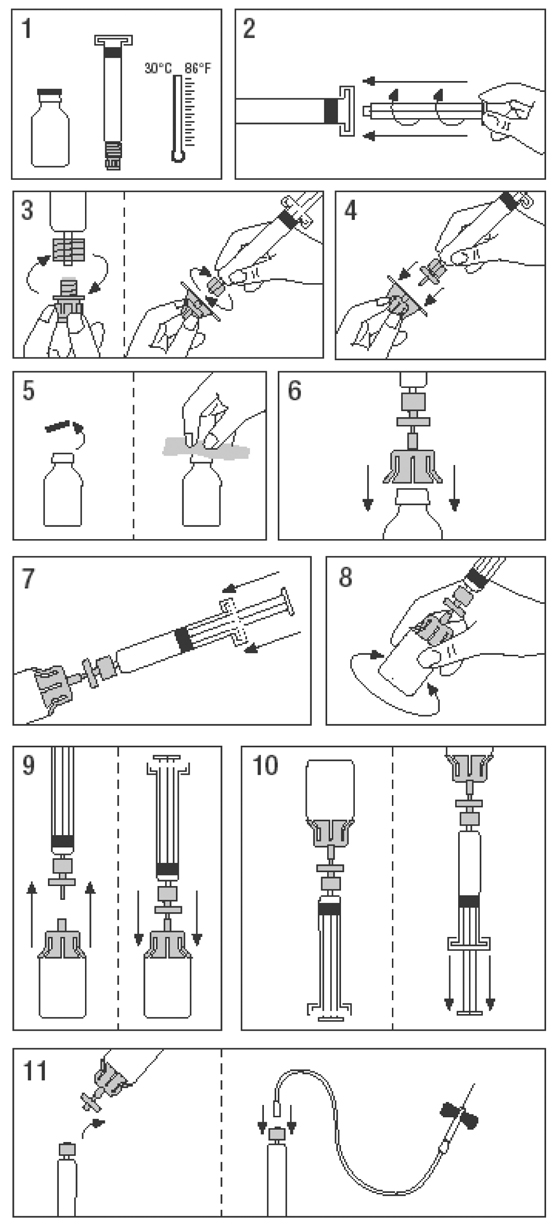
Sobredosificación.
No se han notificado casos de sobredosificación con Alphanate.
Presentación.
Alphanate se presenta en forma de polvo liofilizado estéril, en frascos-ampolla de dosis única, que contienen 250, 500, 1000 o 1500 U.I. de Factor VIII. Alphanate contiene también actividad del Factor de von Willebrand:Cofactor de Ristocetina no inferior a 400 U.I. por cada 1000 U.I. de actividad de factor VIII. La actividad del Factor de von Willebrand:Cofactor de Ristocetina (FVW:RCo) se indica en unidades internacionales (U.I.) en la etiqueta del producto. Alphanate está envasado con una jeringa precargada con disolvente con el volumen adecuado en función de la potencia de Factor Antihemofílico (Agua Estéril para Inyección; 5 ml para las potencias de 250 U.I. y 500 U.I. y 10 ml para las potencias de 1000 U.I. y 1500 U.I.) y accesorios para inyección.