ATRIPLA®
GADOR CHILE
ATRIPLA® es una asociación de dosis fijas de los antivíricos efavirenz, emtricitabina y fumarato de disoproxilo de tenofovir.
Composición.
Cada comprimido contiene: Efavirenz 600 mg, Emtricitabina 200 mg, Fumarato de Disoproxilo de Tenofovir (Equivalente a 245 mg de Disoproxilo de Tenofovir) 300 mg.
Propiedades.
Mecanismo de acción: Efavirenz: El efavirenz es un inhibidor no nucleosídico de la retrotranscriptasa (RT) del VIH-1. La actividad del efavirenz está mediada predominantemente por la inhibición no competitiva de la retrotranscriptasa del VIH-1. El efavirenz no inhibe la retrotranscriptasa del VIH-2 ni las polimerasas a, b, c y d del ADN de las células humanas. Emtricitabina: La emtricitabina, un análogo nucleosídico sintético de la citidina, es fosforilada por las enzimas celulares para formar 5'-trifosfato de emtricitabina. El 5'-trifosfato de emtricitabina inhibe la actividad de la retrotranscriptasa del VIH-1, al competir con el sustrato natural 5'-trifosfato de desoxicitidina e incorporarse en el ADN vírico incipiente, lo que produce la terminación de la cadena. El 5'-trifosfato de emtricitabina es un inhibidor débil de las polimerasas, y del ADN de los mamíferos, y de la polimerasa del ADN mitocondrial. Fumarato de disoproxilo de tenofovir: El fumarato de disoproxilo de tenofovir es un análogo diéster de fosfonato nucleosídico acíclico del monofosfato de adenosina. El FD tenofovir requiere la hidrólisis inicial del diéster para su conversión en tenofovir y fosforilaciones posteriores por las enzimas celulares para formar el difosfato de tenofovir. El difosfato de tenofovir inhibe la actividad de la retrotranscriptasa del VIH-1, al competir con el sustrato natural 5'-trifosfato de desoxiadenosina y, después de su incorporación en el ADN, al finalizar la cadena de ADN. El difosfato de tenofovir es un inhibidor débil de las polimerasas a y b del ADN de los mamíferos, y de la polimerasa c del ADN mitocondrial. Actividad antivírica: Efavirenz, emtricitabina y fumarato de disoproxilo de tenofovir: En estudios de asociaciones en los que se evaluó la actividad antivírica en cultivos de células de la emtricitabina y el efavirenz juntos, el efavirenz y el tenofovir juntos, y la emtricitabina y el tenofovir juntos, se observaron efectos antivíricos entre aditivos y sinérgicos. Efavirenz: La concentración de efavirenz que inhibe la replicación de las cepas no mutantes adaptadas en el laboratorio y cepas clínicas en cultivos de células entre el 90 y el 95% (CE90-95) varió entre 1.7 y 25 nM en cultivos de líneas celulares linfoblastoides, leucocitos mononucleares en la sangre periférica y macrófagos o monocitos. El efavirenz demostró una actividad antivírica aditiva contra el VIH-1, en cultivos de células, al asociarse a inhibidores no nucleosídicos de la retrotranscriptasa (INNRT) (delavirdina y nevirapina), inhibidores nucleosídicos de la retrotranscriptasa (INRT) (abacavir, didanosina, lamivudina, estavudina, zalcitabina y zidovudina), inhibidores de la proteasa (IP) (amprenavir, indinavir, lopinavir, nelfinavir, ritonavir y saquinavir), y el inhibidor de la fusión, enfuvirtida. El efavirenz demostró una actividad antivírica entre aditiva y antagonista en cultivo de células con el atazanavir. El efavirenz demostró una actividad antivírica contra las cepas aisladas B clade y la mayoría de las cepas aisladas B no clade (subtipos A, AE, AG, C, D, F, G, J y N), pero tuvo una actividad antivírica reducida contra los virus del grupo O. El efavirenz no es activo contra el VIH-2. Emtricitabina: Se evaluó la actividad antivírica de la emtricitabina en cultivo de células, contra cepas aisladas clínicas y de laboratorio del VIH-1, en líneas celulares linfoblastoides, la línea de células MAGI-CCR5 y leucocitos mononucleares en la sangre periférica. Los valores de la concentración eficaz al 50% (CE50) de la emtricitabina estuvieron entre los límites de 0.0013 y 0.64 mM (0.0003 a 0.158 mg/ml). En estudios de asociaciones de emtricitabina con inhibidores nucleosídicos de la retrotranscriptasa (abacavir, lamivudina, estavudina, zalcitabina y zidovudina), inhibidores no nucleosídicos de la retrotranscriptasa (delavirdina, efavirenz y nevirapina) e inhibidores de la proteasa (amprenavir, nelfinavir, ritonavir y saquinavir) se observaron efectos entre aditivos y sinérgicos. La emtricitabina presentó actividad antivírica en cultivo de células contra los clades A, B, C, D, E, F y G del VIH-1 (los valores de CE50 estuvieron entre los límites de 0.007 y 0.075 mM) y mostró una actividad específica de las cepas contra el VIH-2 (los valores de CE50 estuvieron entre los límites de 0.007 y 1.5 mM). Fumarato de disoproxilo de tenofovir: Se evaluó la actividad antivírica en cultivo de células del tenofovir contra cepas clínicas y de laboratorio del VIH-1 en las líneas celulares linfoblastoides, células primarias de monocitos/macrófagos y linfocitos en la sangre periférica. Los valores de CE50 correspondientes al tenofovir estuvieron dentro de los límites de 0.04 y 8.5 mM. En estudios de asociaciones farmacológicas del tenofovir con inhibidores nucleosídicos de la retrotranscriptasa (abacavir, didanosina, lamivudina, estavudina, zalcitabina y zidovudina), inhibidores no nucleosídicos de la retrotranscriptasa (delavirdina, efavirenz y nevirapina) e inhibidores de la proteasa (amprenavir, indinavir, nelfinavir, ritonavir y saquinavir) se observaron efectos entre aditivos y sinérgicos. El tenofovir presentó actividad antivírica en cultivo de células contra los clades A, B, C, D, E, F, G y O del VIH-1 (los valores de CE50 variaron entre 0.5 y 2.2 mM), y actividad específica de cepas contra el VIH-2 (los valores de CE50 variaron entre 1.6 mM y 5.5 mM). Resistencia: Efavirenz, emtricitabina y fumarato de disoproxilo de tenofovir: Se han seleccionado en cultivos de células y en estudios clínicos cepas del VIH-1 con una disminución de la sensibilidad a la asociación de emtricitabina y tenofovir. El análisis genotípico de estas cepas aisladas identificó las sustituciones de aminoácidos M184I/V o K65R en la RT vírica. En un estudio clínico con pacientes sin tratamiento antirretrovírico previo [estudio 934, véase Estudios clínicos] se realizó un análisis de resistencia en cepas aisladas del VIH-1 de todos los pacientes con fracaso virológico confirmado con > 400 copias/ml de ARN del VIH-1 en la semana 144 o que habían abandonado prematuramente el estudio. La forma más frecuente de resistencia presente que se produjo fue la resistencia genotípica al efavirenz, predominantemente la sustitución K103N. La resistencia al efavirenz se produjo en 13/19 pacientes analizados del grupo tratado con emtricitabina + FD tenofovir, y en 21/29 pacientes analizados del grupo que recibió la asociación de dosis fijas de zidovudina y lamivudina. Se observó la sustitución de aminoácidos M184V, asociada con resistencia a la emtricitabina y la lamivudina, en 2/19 de las cepas aisladas de los pacientes analizados del grupo tratado con emtricitabina + FD tenofovir y en 10/29 de las cepas aisladas de los pacientes analizados del grupo tratado con la asociación de zidovudina y lamivudina. En las 144 semanas del estudio 934, ningún paciente presentó una sustitución K65R detectable en su VIH-1, según los análisis genotípicos habituales. En un estudio clínico en pacientes sin tratamiento antirretrovírico previo, las cepas aisladas de 8/47 (17%) pacientes analizados, que recibían FD tenofovir, presentaron la mutación K65R hasta las 144 semanas de tratamiento; siete de estos casos se produjeron en las 48 primeras semanas de tratamiento y uno en la semana 96. En los pacientes con tratamiento antirretrovírico previo, 14/304 (5%) de los pacientes tratados con FD tenofovir y con fracaso virológico hasta la semana 96 presentaron una disminución de la sensibilidad > 1.4 vez (mediana, 2.7) al tenofovir. El análisis genotípico de los aislados resistentes mostró una sustitución en el gen de la RT del VIH-1 que produjo la sustitución del aminoácido K65R. En un estudio clínico en pacientes con tratamiento antirretrovírico previo [estudio 073, véase Estudios clínicos] se dispone de datos limitados (N = 3) de pacientes que estaban estables, con supresión virológica y que no tenían antecedentes de fracaso virológico. Los perfiles de resistencia observados en este estudio fueron similares a los del estudio 934. Efavirenz: Se han obtenido cepas aisladas clínicas con una disminución de la sensibilidad al efavirenz en cultivo de células. La sustitución de aminoácido observada con mayor frecuencia en los estudios clínicos con efavirenz es K103N (54%). En pacientes que no respondieron al tratamiento con efavirenz asociado a otros antirretrovíricos se observaron una o más sustituciones de la retrotranscriptasa en las posiciones de los aminoácidos 98, 100, 101, 103, 106, 108, 188, 190, 225, 227 y 230. Otras sustituciones de resistencia que se observó que aparecieron con frecuencia son L100I (7%), K101E/Q/R (14%), V108I (11%), G190S/T/A (7%), P225H (18%) y M230I/L (11%). En una selección en cultivo de células, aparecieron rápidamente cepas aisladas del VIH-1 con una disminución de la sensibilidad al efavirenz (aumento de > 380 veces del valor de CE90). La caracterización genotípica de estos virus identificó sustituciones que produjeron sustituciones de un solo aminoácido L100I o V179D, sustituciones dobles L100I/V108I y sustituciones triples L100I/V179D/Y181C de la retrotranscriptasa. Emtricitabina: Se han seleccionado cepas aisladas del VIH-1 resistentes a la emtricitabina en cultivos de células y en estudios clínicos. El análisis genotípico de estas cepas aisladas demostró que la disminución de la sensibilidad a la emtricitabina se asoció a una sustitución del gen de la retrotranscriptasa del VIH-1 en el codón 184, que produjo una sustitución del aminoácido metionina por valina o isoleucina (M184V/I). Fumarato de disoproxilo de tenofovir: En un cultivo de células se han seleccionado cepas aisladas del VIH-1 con una disminución de la sensibilidad al tenofovir. Estos virus expresaron una sustitución K65R en la retrotranscriptasa y mostraron una reducción del doble al cuádruple de la sensibilidad al tenofovir. Resistencia cruzada: Efavirenz, emtricitabina y fumarato de disoproxilo de tenofovir: Se ha reconocido la resistencia cruzada entre inhibidores no nucleosídicos de la retrotranscriptasa. Se ha reconocido también la resistencia cruzada entre ciertos inhibidores nucleosídicos de la retrotranscriptasa. Las sustituciones M184V/I o K65R seleccionadas en el cultivo de células por la asociación de emtricitabina y tenofovir también se observan en algunas cepas aisladas del VIH-1 de pacientes que no responden al tratamiento con tenofovir en asociación con lamivudina o emtricitabina, y abacavir o didanosina. Por lo tanto, la resistencia cruzada entre estos fármacos puede presentarse en los pacientes cuyos virus hospedan alguna o ambas de estas sustituciones de aminoácidos. Efavirenz: Las cepas aisladas clínicas que se caracterizaron anteriormente como resistentes al efavirenz también fueron fenotípicamente resistentes, en el cultivo de células, a la delavirdina y la venirapina, en comparación con los valores iniciales. Los aislados víricos clínicos, resistentes a la delavirdina o a la nevirapina, con sustituciones asociadas a la resistencia a los INNRT (A98G, L100I, K101E/P, K103N/S, V106A, Y181X, Y188X, G190X, P225H, F227L o M230L) mostraron una disminución de la sensibilidad al efavirenz en el cultivo de células. Más del 90% de las cepas aisladas resistentes a los INRT examinadas en el cultivo de células conservaron la sensibilidad al efavirenz. Emtricitabina: Las cepas aisladas resistentes a la emtricitabina (M184V/I) presentaron una resistencia cruzada a la lamivudina y la zalcitabina; sin embargo, en el cultivo de células, conservaron la sensibilidad a la didanosina, la estavudina, el tenofovir, la zidovudina y los INNRT (delavirdina, efavirenz y nevirapina). Las cepas aisladas de VIH-1 que contienen la sustitución K65R, seleccionados in vivo por el abacavir, la didanosina, el tenofovir y la zalcitabina, presentaron una disminución de la sensibilidad a la inhibición por la emtricitabina. Los virus que hospedan sustituciones que confieren una disminución de la sensibilidad a la estavudina y la zidovudina (M41L, D67N, K70R, L210W, T215Y/F y K219Q/E), o a la didanosina (L74V) se mantuvieron sensibles a la emtricitabina. Fumarato de disoproxilo de tenofovir: La sustitución K65R seleccionada por el tenofovir también se selecciona en algunos individuos infectados por el VIH-1, tratados con abacavir, didanosina o zalcitabina. Las cepas aisladas del VIH-1 con esta sustitución también mostraron una disminución de la sensibilidad a la emtricitabina y la lamivudina. Por lo tanto, puede presentarse la resistencia cruzada en estos fármacos en los pacientes cuyos virus hospedan la mutación K65R. Las cepas aisladas del VIH-1 de pacientes (n = 20) cuyo VIH-1 expresó una media de tres sustituciones de aminoácidos de la RT asociadas a la zidovudina (M41L, D67N, K70R, L210W, T215Y/F o K219Q/E/N) presentaron una disminución de 3.1 veces de la sensibilidad al tenofovir. El VIH-1 resistente a varios nucleosídicos con una sustitución de doble inserción T69S en la RT presentó una disminución de la sensibilidad al tenofovir. En los pacientes cuyo virus expresó una sustitución L74V sin sustituciones asociadas a la resistencia de la zidovudina (N = 8) se observó una disminución de la respuesta a VIREAD. Se dispone de datos limitados en los pacientes cuyo virus expresó una sustitución Y115F (N = 3), una sustitución Q151M (N = 2) o una inserción T69 (N = 4), en todos los cuales se observó una disminución de la respuesta.
Farmacología.
ATRIPLA® es una asociación de dosis fijas, en comprimidos, que contiene efavirenz, emtricitabina y fumarato de disoproxilo de tenofovir (FD tenofovir). STOCRIN es el nombre comercial de efavirenz, un inhibidor no nucleosídico de la retrotranscriptasa. EMTRIVA es el nombre comercial de emtricitabina, un análogo nucleosídico sintético de la citidina. VIREAD es el nombre comercial del FD tenofovir, que se convierte in vivo en tenofovir, un análogo fosfonato nucleosídico acíclico (nucleotídico) del 5'-monofosfato de adenosina. VIREAD y EMTRIVA son los componentes de TRUVADA. Los comprimidos de ATRIPLA® (Efavirenz - Emtricitabina - Fumarato de disoproxilo de tenofovir) son para administración oral. Efavirenz: El nombre químico del efavirenz es (S)-6-cloro-4-(ciclopropiletinil)-1,4-dihidro-4-(trifluorometil)-2H-3,1-benzoxazin-2-ona. Su fórmula molecular es C14H9ClF3NO2 y su fórmula estructural es:

El efavirenz es un polvo cristalino, de color entre blanco y ligeramente rosado, con una masa molecular de 315.68. Es prácticamente no hidrosoluble ( < 10 mg/ml). Emtricitabina: El nombre químico de la emtricitabina es 5-fluoro-1-(2R,5S)-[2-(hidroximetil)-1,3-oxatiolan-5-il]citosina. La emtricitabina es el enantiómero (-) de un análogo tio de la citidina, que difiere de otros análogos de la citidina porque tiene un flúor en la posición 5. Su fórmula molecular es C8H10FN3O3S y su peso molecular es 247.24. Tiene la siguiente fórmula estructural:

La emtricitabina es un polvo cristalino, de color entre blanco y blanquecino, con una hidrosolubilidad de aproximadamente 112 mg/ml, a 25°C. Fumarato de disoproxilo de tenofovir: El fumarato de disoproxilo de tenofovir es una sal del ácido fumárico del derivado éster bis-isopropoxicarboniloximetil del tenofovir. El nombre químico del fumarato de disoproxilo de tenofovir es fumarato de 9-[(R)-2-[[bis[[(isopropoxicarbonil)oxi]-metoxi]fosfinil]metoxi]propil]adenina (1:1). Su fórmula molecular es C19H30N5O10P C4H4O4 y su peso molecular es 635.52. Tiene la siguiente fórmula estructural:
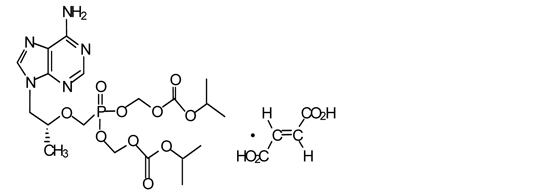
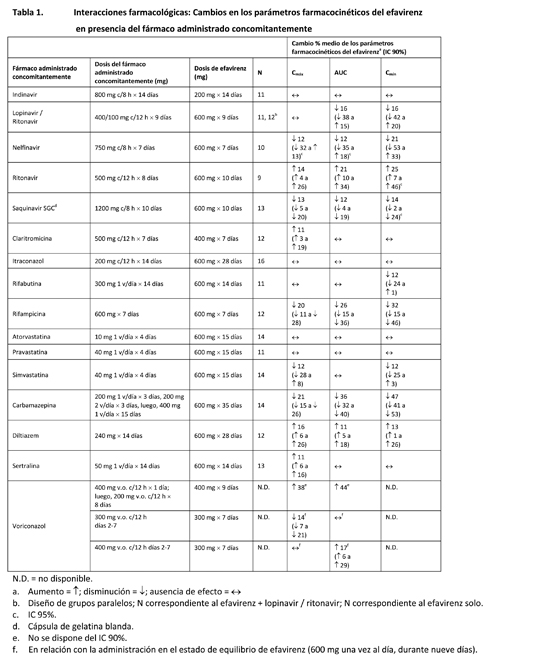
El fumarato de disoproxilo de tenofovir es un polvo cristalino, de color entre blanco y blanquecino, con una hidrosolubilidad de aproximadamente 13.4 mg/ml a 25°C. Farmacología: Farmacocinética: ATRIPLA®: Un comprimido de ATRIPLA® es bioequivalente a un comprimido de STOCRIN (600 mg) más una cápsula de EMTRIVA (200 mg) más un comprimido de VIREAD (300 mg) después de su administración como dosis única a sujetos sanos en ayunas (n = 45). Efavirenz: En los pacientes infectados por el VIH-1, el tiempo hasta alcanzar las concentraciones máximas en el plasma fue de aproximadamente tres a cinco horas, y las concentraciones plasmáticas en el estado de equilibrio se alcanzaron después de seis a diez días. En 35 pacientes infectados por el VIH-1 que recibieron 600 mg de efavirenz una vez al día, la Cmáx en el estado de equilibrio fue de 12.9 ± 3.7 mM (media ± d.e.); la Cmín fue de 5.6 ± 3.2 mM, y el AUC, 184 ± 73 mM·h. El efavirenz presenta una elevada fijación a las proteínas del plasma humano (aproximadamente entre el 99.5 y el 99.75%), predominantemente a la albúmina. Después de la administración de efavirenz marcado con C14, se recuperó del 14 al 34% de la dosis en la orina (principalmente como metabolitos), y se recuperó entre el 16 y el 61% en las heces (principalmente como el fármaco original). En los estudios in vitro se sugiere que las principales isoenzimas responsables del metabolismo del efavirenz son el CYP3A y el CYP2B6. Se ha demostrado que el efavirenz induce las enzimas de CYP, lo que produce la inducción de su propio metabolismo. El efavirenz tiene una vida media terminal de 52 a 76 horas después de la administración de dosis únicas, y de 40 a 55 horas después de la administración de varias dosis. Emtricitabina: La emtricitabina se absorbe de manera rápida después de la administración por vía oral, y se alcanzan concentraciones plasmáticas máximas de una a dos horas después de la dosis. Después de la administración por vía oral de varias dosis de emtricitabina a 20 pacientes infectados por el VIH-1, la concentración máxima (Cmáx) de emtricitabina en el plasma, en el estado de equilibrio, fue de 1.8 ± 0.7 mg/ml (media ± d.e.), y el AUC durante un intervalo de dosificación de 24 horas fue de 10.0 ± 3.1 mg·h/ml. La concentración mínima media en el plasma, en el estado de equilibrio, 24 horas después de administrada la dosis, fue de 0.09 mg/ml. La biodisponibilidad absoluta media de la emtricitabina fue del 93%. La unión in vitro de la emtricitabina a las proteínas plasmáticas en los seres humanos es < 4% y no es dependiente de la concentración, entre los límites de 0.02 y 200 mg/ml. Después de la administración de emtricitabina radiomarcada, se recupera aproximadamente el 86% en la orina y el 13% como metabolitos. Los metabolitos de la emtricitabina son 3'-sulfóxido diastereómeros y su conjugado con ácido glucurónico. La emtricitabina se elimina mediante una combinación de filtración glomerular y secreción tubular activa, con una depuración renal en los adultos con una función renal normal de 213 ± 89 ml/min (media ± d.e.). Después de una dosis única por vía oral, la vida media plasmática de la emtricitabina es de aproximadamente 10 horas. Fumarato de disoproxilo de tenofovir: Después de la administración por vía oral de una dosis única de 300 mg de FD tenofovir a pacientes infectados por el VIH-1 en ayunas, las concentraciones séricas máximas (Cmáx) se alcanzaron en 1.0 ± 0.4 horas (media ± d.e.). Los valores de la Cmáx y el AUC fueron de 296 ± 90 ng/ml y 2287 ± 685 ng•h/ml, respectivamente. La biodisponibilidad oral del tenofovir a partir del FD tenofovir en los pacientes en ayunas es de aproximadamente el 25%. La unión in vitro del tenofovir a las proteínas plasmáticas en los seres humanos es < 0.7% y es independiente de la concentración, dentro de los límites de 0.01 y 25 mg/ml. Aproximadamente entre el 70 y el 80% de la dosis por vía intravenosa de tenofovir se recupera como fármaco inalterado en la orina. El tenofovir se elimina mediante una combinación de filtración glomerular y secreción tubular activa, con una depuración renal en los adultos con una función renal normal de 243 ± 33 ml/min (media ± d.e.). Después de una dosis oral única de tenofovir, la vida media de eliminación terminal es de aproximadamente 17 horas. Efectos de los alimentos en la absorción oral: No se ha evaluado ATRIPLA® en presencia de alimentos. La administración de comprimidos de efavirenz con una comida rica en grasas aumentó el AUC media del efavirenz en un 28%, y la Cmáx media, en un 79%, en comparación con su administración en ayunas. En comparación con la administración en ayunas, la administración de una asociación de FD tenofovir y emtricitabina, con una comida rica en grasas o una comida ligera, aumentó el AUC media en un 35%, y la Cmáx, en un 15%, sin ningún efecto sobre la exposición a la emtricitabina [véase Dosificación y Formas de dosificación y concentraciones]. Poblaciones especiales: Raza: Efavirenz: Se aprecia que las características farmacocinéticas del efavirenz en los pacientes infectados con el VIH-1 son similares entre los grupos raciales estudiados. Emtricitabina: No se han identificado diferencias farmacocinéticas debidas a la raza después de la administración de emtricitabina. Fumarato de disoproxilo de tenofovir: No hubo un número suficiente de sujetos de grupos raciales y étnicos, aparte de la raza blanca, para poder determinar adecuadamente las posibles diferencias farmacocinéticas entre estas poblaciones después de la administración de FD tenofovir. Sexo: Efavirenz, emtricitabina y fumarato de disoproxilo de tenofovir: Las propiedades farmacocinéticas del efavirenz, la emtricitabina y el tenofovir son parecidas en los pacientes de ambos sexos. Pacientes pediátricos y geriátricos: No se han realizado estudios farmacocinéticos del FD tenofovir en los pacientes pediátricos ( < 18 años). No se ha estudiado el efavirenz en los pacientes pediátricos menores de tres años o con un peso inferior a 13 kg. Se ha estudiado la emtricitabina en los pacientes pediátricos de tres meses a 17 años. No se recomienda la administración de ATRIPLA® a los pacientes pediátricos. No se han evaluado completamente las características farmacocinéticas del efavirenz, la emtricitabina y el tenofovir en los ancianos (≥ 65 años). [véase Uso en poblaciones específicas]. Pacientes con disfunción renal: Efavirenz: No se han estudiado las característica farmacocinéticas del efavirenz en los pacientes con insuficiencia renal; sin embargo, menos del 1% del efavirenz se excreta inalterado por la orina, por lo que la repercusión de la disfunción renal sobre la eliminación del efavirenz debería ser mínima. Emtricitabina y fumarato de disoproxilo de tenofovir: Las propiedades farmacocinéticas de la emtricitabina y del FD tenofovir están alteradas en los pacientes con disfunción renal. En los pacientes con una depuración de creatinina < 50 ml/min, la Cmáx y el AUC0-inf de la emtricitabina y el tenofovir aumentaron [véase Advertencias y Precauciones]. Pacientes con disfunción hepática: Efavirenz: No se han estudiado adecuadamente las características farmacocinéticas del efavirenz en los pacientes con disfunción hepática [véase Advertencias y Precauciones]. Emtricitabina: No se han estudiado las características farmacocinéticas de la emtricitabina en los pacientes con disfunción hepática; sin embargo, las enzimas hepáticas no metabolizan significativamente la emtricitabina, por lo que la repercusión de la disfunción hepática debería ser limitada. Fumarato de disoproxilo de tenofovir: Se han estudiado las características farmacocinéticas del tenofovir después de una dosis de 300 mg de FD tenofovir en pacientes no infectados por el VIH y con disfunción hepática de moderada a grave. No hubo alteraciones importantes en las propiedades farmacocinéticas del tenofovir en pacientes con disfunción hepática, en comparación con los pacientes con una función hepática normal. Evaluación de las interacciones farmacológicas: Los estudios de interacción farmacológica descritos se efectuaron con efavirenz, emtricitabina o FD tenofovir como fármacos individuales; no se han realizado estudios de interacción farmacológica con ATRIPLA®. Efavirenz: Las características farmacocinéticas del efavirenz y el tenofovir en el estado de equilibrio no se vieron afectadas cuando se administraron efavirenz y fumarato de disoproxilo de tenofovir juntos, en comparación con la administración de cada fármaco por separado. No se han realizado estudios específicos de interacción farmacológica con efavirenz e INRT aparte del tenofovir, la lamivudina y la zidovudina. No se espera que se produzcan interacciones clínicamente significativas basadas en las vías de eliminación de los INRT. Se ha demostrado que el efavirenz causa, in vivo, inducción de las enzimas hepáticas, aumentando así la biotransformación de algunos fármacos metabolizados por el CYP3A. En estudios in vitro se ha demostrado que el efavirenz inhibió las isoenzimas 2C9, 2C19 y 3A4 delCYP, con valores de Ki (entre 8.5 y 17 mM) dentro de los límites de las concentraciones plasmáticas observadas de efavirenz. En los estudios in vitro, el efavirenz no inhibió el CYP2E1, e inhibió el CYP2D6 y el CYP1A2 (valores de Ki entre 82 y 160 mM), sólo a concentraciones muy superiores a las alcanzadas clínicamente. La administración concomitante de efavirenz con fármacos que son metabolizados principalmente por las isoenzimas 2C9, 2C19 y 3A4 puede producir una alteración de las concentraciones plasmáticas del fármaco administrado concomitantemente. Se esperaría que los fármacos que inducen la actividad del CYP3A4 aumenten la depuración del efavirenz, produciéndose una disminución de las concentraciones plasmáticas. Se realizaron estudios de interacción farmacológica con efavirenz y otros fármacos con probabilidad de ser administrados concomitantemente o con fármacos usados con frecuencia como sondas para la interacción farmacocinética. No se observó ninguna interacción clínicamente significativa entre el efavirenz y la zidovudina, la lamivudina, la azitromicina, el fluconazol, el lorazepam, la cetirizina ni la paroxetina. Las dosis únicas de famotidina o de un antiácido de aluminio y magnesio con simeticona no tuvieron ningún efecto sobre las exposiciones del efavirenz. Los efectos de la administración concomitante de efavirenz sobre la Cmáx, el AUC y la Cmín se resumen en la tabla 1 (efecto de otros fármacos sobre el efavirenz) y en la tabla 2 (efecto del efavirenz sobre otros fármacos). Puede consultarse más información acerca de las recomendaciones clínicas en Interacciones.
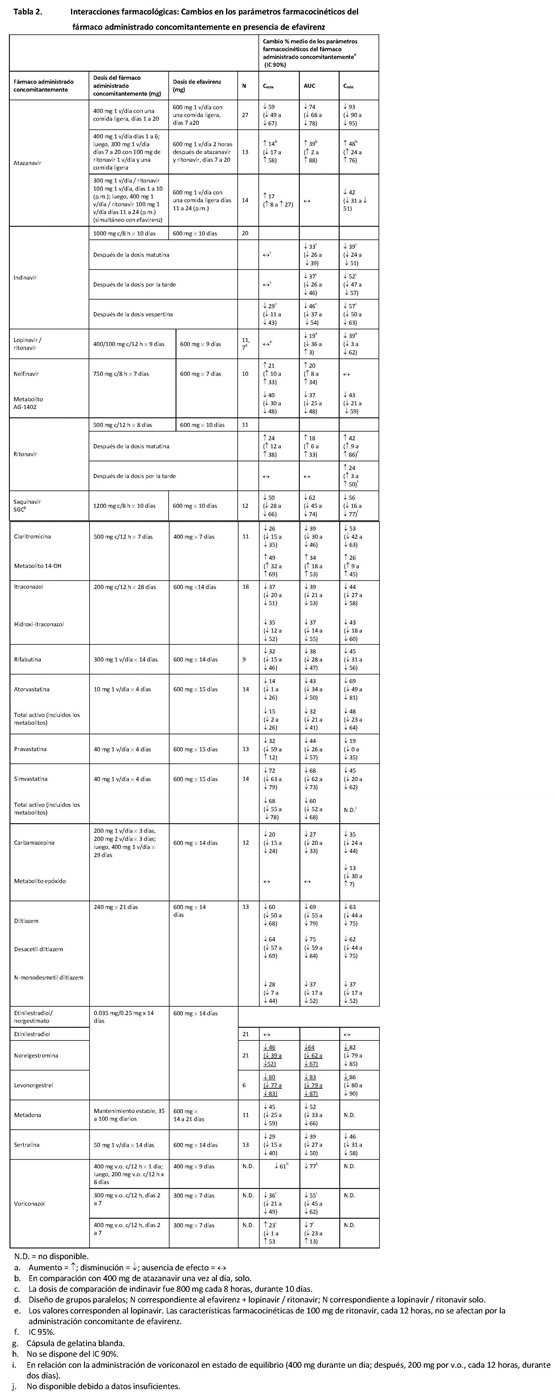
Emtricitabina y fumarato de disoproxilo de tenofovir: Las características farmacocinéticas de la emtricitabina y del tenofovir en el estado de equilibrio no se vieron afectadas cuando se administraron emtricitabina y fumarato de disoproxilo de tenofovir juntos, en comparación con la administración de cada fármaco por separado. En los estudios de interacción farmacológica in vitro y en las propiedades farmacocinéticas clínicas se ha demostrado que la posibilidad de interacciones mediadas por el CYP que afectan a la emtricitabina y al tenofovir con otros medicamentos es baja. La emtricitabina y el tenofovir se excretan principalmente por los riñones, mediante una combinación de filtración glomerular y secreción tubular activa. No se observaron interacciones farmacológicas debido a la competencia por la excreción renal. Sin embargo, la administración de emtricitabina y FD tenofovir concomitantemente con medicamentos que se eliminan por secreción tubular activa puede aumentar las concentraciones de emtricitabina, tenofovir o del fármaco administrado concomitantemente. Los fármacos que disminuyen la función renal también pueden aumentar las concentraciones de emtricitabina o tenofovir. No se observaron interacciones farmacológicas clínicamente significativas entre la emtricitabina y el famciclovir, el indinavir, la estavudina, el fumarato de disoproxilo de tenofovir y la zidovudina. De manera parecida, en estudios realizados en voluntarios sanos, no se observaron interacciones farmacológicas clínicamente significativas entre el fumarato de disoproxilo de tenofovir y el abacavir, el efavirenz, la emtricitabina, el entecavir, el indinavir, la lamivudina, la asociación de lopinavir y ritonavir, la metadona, el nelfinavir, los anticonceptivos orales, la ribavirina, la asociación de saquinavir y ritonavir, o el tacrolimus. Después de administrar varias dosis a pacientes negativos para el VIH que estaban recibiendo tratamiento de mantenimiento crónico con metadona, anticonceptivos orales o dosis únicas de ribavirina, las características farmacocinéticas del tenofovir en el estado de equilibrio fueron parecidas a las observadas en estudios anteriores, lo que indica la ausencia de interacciones farmacológicas clínicamente significativas entre estos fármacos y el FD tenofovir.
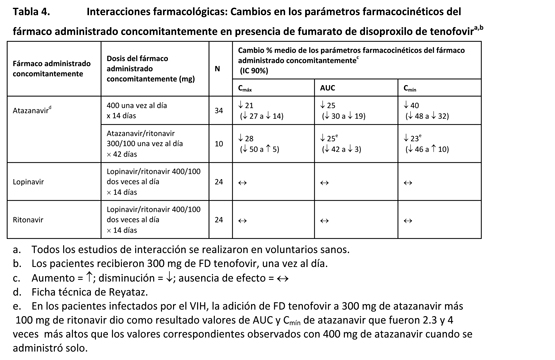
Los efectos de los fármacos administrados concomitantemente sobre la Cmáx, el AUC y la Cmín del tenofovir se muestran en la tabla 3. Los efectos de la administración concomitante de FD tenofovir sobre la Cmáx, el AUC y la Cmín de los fármacos administrados concomitantemente se muestran en las tablas 4 y 5.
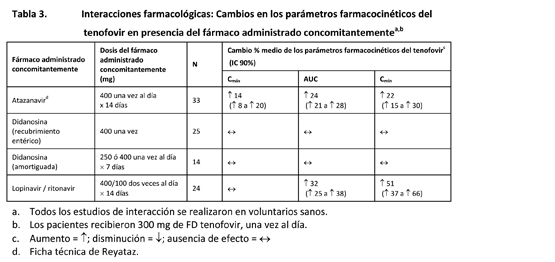
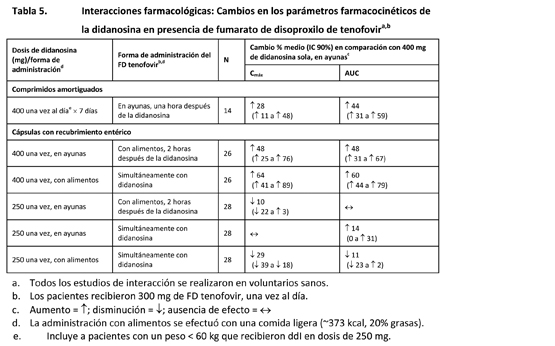
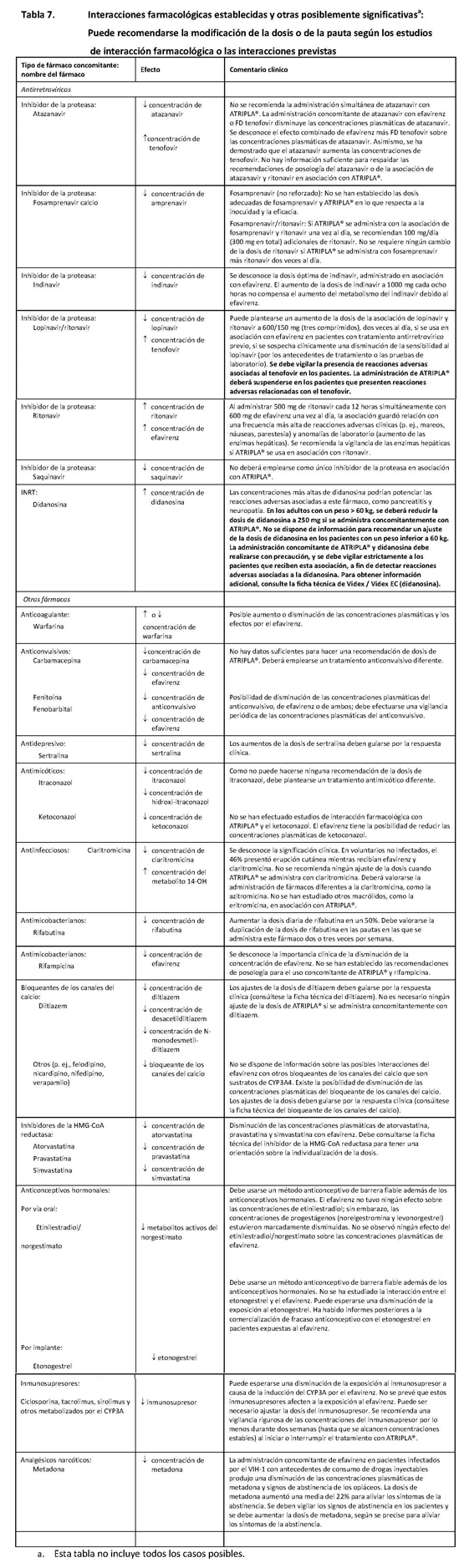
La administración concomitante de fumarato de disoproxilo de tenofovir con didanosina produce cambios en las características farmacocinéticas de la didanosina que pueden tener importancia clínica. En la tabla 5 se resumen los efectos del fumarato de disoproxilo de tenofovir en las características farmacocinéticas de la didanosina. La administración concomitante de fumarato de disoproxilo de tenofovir con comprimidos amortiguados o con cápsulas con recubrimiento entérico de didanosina aumenta significativamente la Cmáx y el AUC de la didanosina. Cuando se administraron cápsulas con recubrimiento entérico de 250 mg de didanosina con fumarato de disoproxilo de tenofovir, las exposiciones sistémicas a la didanosina fueron parecidas a las observadas con las cápsulas con recubrimiento entérico de 400 mg solas, administradas en ayunas. Se desconoce el mecanismo de esta interacción [véanse las recomendaciones de ajuste de la posología de la didanosina en Interacciones, tabla 7].
Indicaciones.
ATRIPLA® está indicado para su uso solo, como pauta completa o en asociación con otros antirretrovíricos, para el tratamiento de la infección por el virus de la inmunodeficiencia humana de tipo 1 (VIH-1) en los adultos.
Dosificación.
Adultos: La dosis de ATRIPLA® es de un comprimido, tomado una vez al día por vía oral, con el estómago vacío. La administración al acostarse puede mejorar la tolerabilidad de los síntomas del sistema nervioso. Uso pediátrico: No se recomienda el uso de ATRIPLA® en los pacientes menores de 18 años, porque es un comprimido de asociación de dosis fijas que contiene un componente, el fumarato de disoproxilo de tenofovir, cuya inocuidad y eficacia no se han establecido en este grupo de edad. Uso geriátrico: En los estudios clínicos de efavirenz, emtricitabina o FD tenofovir no se incluyó un número suficiente de pacientes con una edad a partir de 65 años como para determinar si responden de manera diferente a los pacientes más jóvenes. En general, la selección de la dosis para los pacientes ancianos debe ser cautelosa y se debe tener en cuenta la mayor frecuencia de disminución de las funciones cardiaca, renal o hepática, y las enfermedades concomitantes u otros tratamientos con fármacos. Ajuste de la dosis en caso de disfunción renal: Dado que ATRIPLA® es una asociación de dosis fijas, no deberá recetarse a los pacientes que precisan un ajuste de la dosificación, por ejemplo, los que sufren una disfunción renal moderada o grave (depuración de creatinina < 50 ml/min). Formas de dosificación y concentraciones: ATRIPLA® se presenta en comprimidos. Cada comprimido contiene 600 mg de efavirenz, 200 mg de emtricitabina y 300 mg de fumarato de disoproxilo de tenofovir (FD tenofovir, que es equivalente a 245 mg de disoproxilo de tenofovir). Los comprimidos son rosados, en forma de cápsula, recubiertos con película, grabados con "123" en una cara y lisos en la otra.
Contraindicaciones.
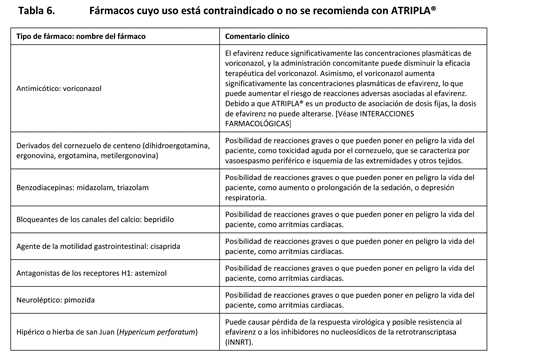
ATRIPLA® está contraindicado en los pacientes con hipersensibilidad clínicamente significativa, previamente demostrada (por ejemplo, síndrome de Stevens-Johnson, eritema multiforme o erupciones cutáneas tóxicas) al efavirenz, un componente de ATRIPLA®. Fármacos contraindicados: En el caso de algunos fármacos, la competencia por el CYP3A por el efavirenz podría causar la inhibición de su metabolismo y crear la posibilidad de reacciones adversas graves o que pueden poner en peligro la vida del paciente (por ejemplo, arritmias cardiacas, sedación prolongada o depresión respiratoria). En la tabla 6 se enumeran los fármacos que están contraindicados con ATRIPLA®.
Reacciones adversas.
Efavirenz, emtricitabina y fumarato de disoproxilo de tenofovir: en otros apartados de la ficha técnica se tratan las siguientes reacciones adversas: Acidosis láctica o hepatomegalia grave con esteatosis [Advertencias]. Exacerbaciones agudas y graves de la hepatitis B [Advertencias]. Síntomas psiquiátricos [Advertencias]. Síntomas del sistema nervioso [Advertencias]. Nueva aparición o empeoramiento de la disfunción renal [Advertencias]. Erupción cutánea [Advertencias]. Disminución de la densidad mineral ósea [Advertencias]. Síndrome de reconstitución inmunitaria [Advertencias]. Interacciones farmacológicas [Contraindicaciones, Advertencias e Interacciones farmacológicas]. Si se desea información adicional acerca de la seguridad de STOCRIN (efavirenz) EMTRIVA (emtricitabina) o VIREAD (FD tenofovir) en asociación con otros antirretrovíricos, consúltese la ficha técnica de estos productos. Reacciones adversas de la experiencia de los ensayos clínicos: Debido a que los ensayos clínicos se realizan en condiciones muy variadas, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no pueden compararse directamente con las tasas de los ensayos clínicos de otro fármaco y puede que no reflejen las tasas observadas en la práctica. Estudio 934: El estudio 934 fue un estudio de diseño abierto y controlado con fármaco activo, en el que 511 pacientes sin tratamiento antirretrovírico previo recibieron emtricitabina + FD tenofovir administrados en asociación con efavirenz (n = 257), o la asociación de zidovudina y lamivudina, en asociación con efavirenz (n = 254). Las reacciones adversas más frecuentes (incidencia ≥ 10%, cualquier gravedad) que se produjeron en el estudio 934 fueron diarrea, náuseas, fatiga, cefalea, mareos, depresión, insomnio, sueños anormales y erupción cutánea. Las reacciones adversas observadas en este estudio fueron, en general, coherentes con las observadas en estudios anteriores de los componentes por separado (tabla 10).
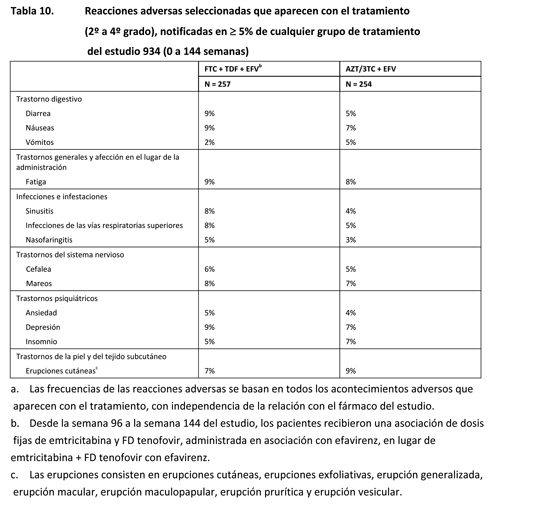
Estudio 073: En el estudio 073, se asignó aleatoriamente a los pacientes con supresión virológica estable, en tratamiento antirretrovírico y sin antecedentes de fracaso virológico, para que recibieran ATRIPLA® o permanecieran en su pauta de tratamiento inicial. Las reacciones adversas observadas en el estudio 073 fueron, en general, coherentes con las observadas en el estudio 934 y con las observadas con los componentes individuales de ATRIPLA® cuando cada uno de ellos se administró asociado a otros antirretrovíricos. Además de las reacciones adversas observadas en los estudios 934 y 073, se observaron las siguientes reacciones adversas en los ensayos clínicos de efavirenz, emtricitabina o FD tenofovir en asociación con otros antirretrovíricos. Efavirenz: Las reacciones adversas más significativas observadas en los pacientes tratados con efavirenz son los síntomas del sistema nervioso (ver Advertencias), los síntomas psiquiátricos (ver Advertencias) y las erupciones cutáneas (ver Advertencias). Algunas reacciones adversas seleccionadas, de intensidad moderada a grave, observadas en ≥ 2% de los pacientes tratados con efavirenz, en dos ensayos clínicos controlados fueron dolor, alteración de la concentración, sueños anormales, somnolencia, anorexia, dispepsia, dolor abdominal, nerviosismo y prurito. Se ha descrito también pancreatitis, aunque no se ha establecido una relación de causalidad con el efavirenz. En un número significativamente más alto de pacientes tratados con 600 mg de efavirenz se observaron aumentos asintomáticos de las concentraciones de amilasa sérica, en comparación con los testigos. Emtricitabina y fumarato de disoproxilo de tenofovir: Las reacciones que se produjeron al menos en el 5% de los pacientes con o sin tratamiento previo con antirretrovíricos, tratados con emtricitabina o FD tenofovir con otros antirretrovíricos en los estudios clínicos, fueron artralgia, aumento de la tos, dispepsia, fiebre, mialgia, dolor, dolor abdominal, dolor lumbar, parestesia, neuropatía periférica (incluidas neuritis periférica y neuropatía), neumonía, rinitis y erupciones cutáneas (incluidas erupción cutánea, prurito, erupción maculopapular, urticaria, erupción vesiculoampollar, erupción pustular y reacción alérgica). En los pacientes tratados con emtricitabina se ha descrito la decoloración de la piel con una frecuencia más alta; se manifestó por hiperpigmentación de las palmas de las manos o las plantas de los pies, y generalmente fue leve y asintomática. Se desconocen su mecanismo y su importancia clínica. Anomalías de laboratorio: Efavirenz, emtricitabina y fumarato de disoproxilo de tenofovir: En general, las anomalías de laboratorio observadas en el estudio 934 (tabla 11) fueron coherentes con las observadas en estudios anteriores.
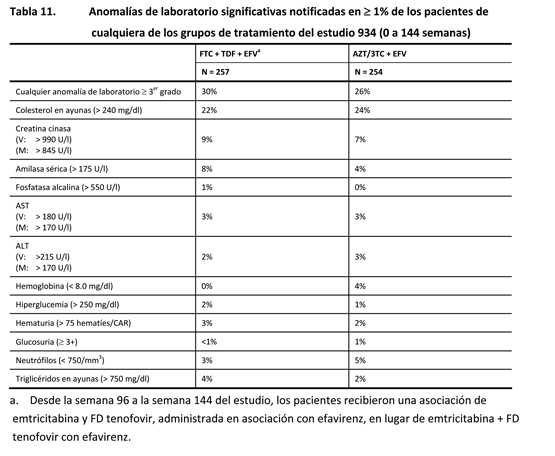
Las anomalías de laboratorio observadas en el estudio 073 fueron, en general, coherentes con las del estudio 934. Además de las anomalías de laboratorio descritas en el estudio 934 (tabla 11), se produjeron aumentos de grado 3/4 en la bilirrubina ( > 2.5 x LSN), la amilasa pancreática ( > 2.0 x LSN), la glucosa sérica ( < 40 ó > 250 mg/dl) y la lipasa sérica ( > 2.0 x
LSN) hasta en el 3% de los pacientes tratados con emtricitabina o FD tenofovir con otros antirretrovíricos en ensayos clínicos. Reacciones adversas hepáticas: En el estudio 934, 19 pacientes tratados con efavirenz, emtricitabina y FD tenofovir, y 20 pacientes tratados con efavirenz y una asociación de dosis fijas de zidovudina y lamivudina fueron positivos para el antígeno de superficie de la hepatitis B o para los anticuerpos de la hepatitis C. De estos pacientes con infección concomitante, un paciente (1/19) del grupo tratado con efavirenz, emtricitabina y FD tenofovir presentó aumentos de las transaminasas de más de cinco veces el valor del LSN (límite superior de la normalidad) hasta las 144 semanas. En el grupo que recibió la asociación de dosis fijas de zidovudina y lamivudina, dos pacientes (2/20) presentaron aumentos de las transaminasas de más de cinco veces el LSN hasta las 144 semanas. Ningún paciente con infección concomitante por el VHB, el VHC o por ambos abandonó el estudio debido a trastornos hepatobiliares [ver Advertencias]. Experiencia posterior a la comercialización: Se han identificado las siguientes reacciones adversas durante el uso posterior a la aprobación del efavirenz, la emtricitabina y el FD tenofovir. Debido a que las reacciones posteriores a la comercialización se notifican voluntariamente a partir de una población de tamaño indeterminado, no siempre es posible calcular de manera fiable su frecuencia ni establecer una relación causal con la exposición al fármaco. Efavirenz: Trastornos cardíacos: Palpitaciones. Trastornos del oído y del laberinto: Tinnitus. Trastornos endocrinos: Ginecomastia. Trastornos oculares: Anomalías de la visión. Trastornos digestivos: Estreñimiento, malabsorción. Trastornos generales y anomalías en el lugar de administración: Astenia. Trastornos hepatobiliares: Aumento de las enzimas hepáticas, insuficiencia hepática, hepatitis. Trastornos del sistema inmunitario: Reacciones alérgicas. Trastornos del metabolismo y de la nutrición: Redistribución o acumulación de la grasa corporal (ver Advertencias). Hipercolesterolemia, hipertrigliceridemia. Trastornos musculoesqueléticos y del tejido conectivo: Artralgia, mialgia, miopatía. Trastornos del sistema nervioso: Anomalías de la coordinación, ataxia, trastornos cerebelosos de la coordinación y el equilibrio, convulsiones, hipoestesia, parestesia, neuropatía, temblores, vértigo. Trastornos psiquiátricos: Reacciones de agitación, agitación, ideas delirantes, labilidad emocional, manía, neurosis, paranoia, psicosis, suicidio. Trastornos respiratorios, torácicos y mediastínicos: Disnea. Trastornos de la piel y del tejido subcutáneo: Rubor, eritema multiforme, dermatitis fotoalérgica, síndrome de Stevens-Johnson. Trastornos vasculares: Rubor (crisis vasomotora). Emtricitabina: No se han identificado reacciones adversas adicionales, posteriores a la comercialización, para su inclusión en este apartado. Fumarato de disoproxilo de tenofovir: Trastornos del sistema inmunitario: Reacción alérgica (incluido el angioedema). Trastornos del metabolismo y de la nutrición: Acidosis láctica, hipopotasemia, hipofosfatemia. Trastornos respiratorios, torácicos y mediastínicos: Disnea. Trastornos digestivos: Pancreatitis, aumento de la amilasa, dolor abdominal. Trastornos hepatobiliares: Esteatosis hepática, hepatitis, aumento de las enzimas hepáticas (con mayor frecuencia, aumento de la AST, la ALT y la c-GT). Trastornos de la piel y del tejido subcutáneo: Erupción cutánea. Trastornos musculoesqueléticos y del tejido conectivo: Rabdomiólisis, osteomalacia (manifestada como dolor óseo y que puede contribuir a fracturas), debilidad muscular, miopatía. Trastornos renales y urinarios: Insuficiencia renal aguda, insuficiencia renal, necrosis tubular aguda, síndrome de Fanconi, tubulopatía renal proximal, nefritis intersticial (incluidos casos agudos), diabetes insípida nefrógena, insuficiencia renal, aumento de la creatinina, proteinuria, poliuria. Trastornos generales y afección en el lugar de la administración: Astenia. Las siguientes reacciones adversas, enumeradas bajo los encabezados de sistemas corporales anteriores, pueden producirse a consecuencia de tubulopatía renal proximal: rabdomiólisis, osteomalacia, hipopotasemia, debilidad muscular, miopatía, hipofosfatemia.
Advertencias.
Acidosis láctica y hepatomegalia grave con esteatosis: Se ha notificado la aparición de acidosis láctica y hepatomegalia grave con esteatosis, incluso casos mortales, con el uso de los análogos nucleosídicos, incluido el FD tenofovir, un componente de ATRIPLA®, en asociación con otros antirretrovíricos. La mayoría de estos casos se registraron en mujeres. La obesidad y la exposición prolongada a los nucleosídicos pueden ser factores de riesgo. Se debe tener especial precaución cuando se administran nucleosídicos a cualquier paciente con factores de riesgo conocidos para las enfermedades hepáticas. Sin embargo, también se han notificado casos en los pacientes que no tenían factores de riesgo conocidos. El tratamiento con ATRIPLA® se deberá interrumpir en cualquier paciente que presente resultados clínicos o de laboratorio que sugieran acidosis láctica o hepatotoxicidad pronunciada (lo cual puede incluir hepatomegalia y esteatosis aun en ausencia de aumentos marcados de la transaminasa). Pacientes con infección concomitante por el VIH-1 y el VHB: Se recomienda que se realice la prueba para detectar la presencia del virus de la hepatitis B crónico a todos los pacientes infectados por el VIH-1 antes de iniciar la terapia antirretrovírica. ATRIPLA® no está aprobado para el tratamiento de la infección crónica por el virus de la hepatitis B (VHB), y no se han establecido su eficacia ni su inocuidad en los pacientes infectados concomitantemente por el VHB y el VIH-1. Se han notificado casos de exacerbación aguda y grave de la hepatitis B en los pacientes infectados concomitantemente por el VHB y el VIH-1 que han suspendido la administración de emtricitabina o FD tenofovir, dos de los componentes de ATRIPLA®. En algunos pacientes infectados por el VHB y tratados con emtricitabina, las exacerbaciones de la hepatitis B se asociaron con descompensación hepática e insuficiencia hepática. Se debe vigilar rigurosamente a los pacientes que estén infectados concomitantemente por el VIH-1 y el VHB, con seguimiento clínico y de laboratorio durante por lo menos varios meses después de interrumpir el tratamiento con ATRIPLA®. Si fuese conveniente, puede estar justificado el inicio del tratamiento contra la hepatitis B. ATRIPLA® no deberá administrarse con HEPSERA® (dipivoxilo de adefovir) [véase Interacciones]. Interacciones farmacológicas: Las concentraciones plasmáticas de efavirenz pueden resultar alteradas por los sustratos, inhibidores o inductores del CYP3A. Asimismo, el efavirenz puede alterar las concentraciones plasmáticas de los fármacos metabolizados por el CYP3A (Contraindicaciones, Fármacos contraindicados). Administración concomitante de productos relacionados: Entre los fármacos relacionados que no deben administrarse concomitantemente con ATRIPLA® se cuentan EMTRIVA (emtricitabina), VIREAD (FD tenofovir), TRUVADA (emtricitabina/FD tenofovir) y STOCRIN (efavirenz), que contienen los mismos principios activos que ATRIPLA®. Debido a las semejanzas entre la emtricitabina y la lamivudina, no se debe administrar ATRIPLA® concomitantemente con otros fármacos que contengan lamivudina, como Combivir (lamivudina y zidovudina), Epivir o Epivir-VHB (lamivudina), Epzicom (sulfato de abacavir y lamivudina), Trizivir (sulfato de abacavir, lamivudina y zidovudina) o Hepsera (dipivoxilo de adefovir). Síntomas psiquiátricos: En los pacientes tratados con efavirenz se han notificado experiencias adversas psiquiátricas graves. En ensayos clínicos controlados de 1008 pacientes con pautas que contienen efavirenz durante una media de 2.1 años y de 635 pacientes tratados con pautas testigos durante una media de 1.5 año, las frecuencias (con independencia de la causalidad) de reacciones psiquiátricas graves y específicas en los pacientes que recibieron efavirenz o pautas testigos, respectivamente, fueron: depresión grave (2.4%, 0.9%), ideación suicida (0.7%, 0.3%), intentos de suicidio no mortales (0.5%, 0%), conducta agresiva (0.4%, 0.5%), reacciones paranoides (0.4%, 0.3%) y reacciones maníacas (0.2%, 0.3%). Cuando los síntomas psiquiátricos similares a los que se acaba de mencionar se combinaron y evaluaron como un grupo, en un análisis multifactorial de datos del estudio AI266006 (006), el tratamiento con efavirenz se asoció a un aumento de la aparición de estos síntomas psiquiátricos selectos. Otros factores asociados a un aumento de la presentación de estos síntomas psiquiátricos fueron: antecedentes de consumo de drogas inyectables, antecedentes psiquiátricos y toma de medicamentos psiquiátricos en la entrada en el estudio. Se observaron relaciones parecidas tanto en el grupo de tratamiento con efavirenz como en el testigo. En el estudio 006, la aparición de nuevos síntomas psiquiátricos graves se produjo durante todo el estudio, tanto en los pacientes tratados con efavirenz como en los que recibieron el tratamiento testigo. El 1% de los pacientes tratados con efavirenz suspendió o interrumpió el tratamiento a causa de uno o más de estos síntomas psiquiátricos selectos. También se han notificado casos ocasionales, después de la comercialización, de suicidio, ideas delirantes y comportamiento de tipo psicótico, aunque a partir de estas notificaciones no puede establecerse una relación de causalidad con el uso de efavirenz. Los pacientes con reacciones adversas psiquiátricas graves deben solicitar inmediatamente una evaluación médica para valorar la posibilidad de que los síntomas estén relacionados con el uso de efavirenz y, de ser así, establecer si los riesgos de la continuación del tratamiento superan a las ventajas [véase Reacciones adversas]. Síntomas del sistema nervioso: El 53% (531/1008) de los pacientes que recibieron efavirenz en ensayos clínicos controlados manifestaron síntomas del sistema nervioso central (de cualquier grado, con independencia de la causalidad), en comparación con el 25% (156/635) de los pacientes que recibieron pautas testigos. Estos síntomas consistieron en mareos (28.1% de los 1008 pacientes), insomnio (16.3%), alteración de la concentración (8.3%), somnolencia (7.0%), sueños anormales (6.2%) y alucinaciones (1.2%). Otros síntomas comunicados fueron euforia, confusión, agitación, amnesia, estupor, pensamientos anormales y despersonalización. La mayoría de estos síntomas fueron leves o moderados (50.7%); los síntomas fueron graves en el 2.0% de los pacientes. En total, como resultado de ello, el 2.1% de los pacientes suspendieron el tratamiento. Estos síntomas comienzan habitualmente durante el primer o el segundo día del tratamiento y, por lo general, se resuelven después de las dos a cuatro primeras semanas de tratamiento. Después de cuatro semanas de tratamiento, la prevalencia de síntomas del sistema nervioso de gravedad por lo menos moderada fue del 5 al 9% en los pacientes tratados con pautas que contenían efavirenz, y del 3 al 5% en los pacientes que recibieron una pauta testigo. Se debe informar a los pacientes de que es probable que estos síntomas frecuentes mejoren con la continuación del tratamiento y no fueron predictivos de la aparición posterior de los síntomas psiquiátricos menos frecuentes. La administración de los fármacos al acostarse puede mejorar la tolerabilidad de estos síntomas del sistema nervioso [véase Dosificación]. El análisis de los datos a largo plazo del estudio 006 (mediana del seguimiento, 180 semanas en los pacientes tratados con efavirenz + zidovudina + lamivudina; 102 semanas en los que recibieron efavirenz + indinavir y 76 semanas en los tratados con indinavir + zidovudina + lamivudina) mostró que, pasadas las 24 semanas de tratamiento, las incidencias de los síntomas del sistema nervioso de nueva aparición en los pacientes tratados con efavirenz fueron generalmente parecidas a las del grupo testigo que contenía indinavir. Debe alertarse a los pacientes que reciben ATRIPLA® acerca de la posibilidad de efectos aditivos del sistema nervioso central cuando ATRIPLA® se usa concomitantemente con alcohol o con fármacos psicoactivos. Los pacientes que sufren síntomas del sistema nervioso central como mareos, alteración de la concentración o somnolencia deberán evitar la realización de tareas posiblemente peligrosas como conducir o usar máquinas. Nueva aparición o empeoramiento de la disfunción renal: La emtricitabina y el tenofovir se eliminan principalmente por los riñones; sin embargo, el efavirenz, no. Puesto que ATRIPLA® es un producto de asociación y la dosis de los componentes individuales no puede modificarse, los pacientes con una depuración de creatinina < 50 ml/min no deberán recibir ATRIPLA®. Se han notificado casos de trastornos renales, entre ellos casos de insuficiencia renal aguda y síndrome de Fanconi (lesión tubular renal con hipofosfatemia grave), asociados con el uso del FD tenofovir [véase Reacciones adversas]. Se recomienda calcular la depuración de creatinina en todos los pacientes antes de iniciar la terapia y según se requiera clínicamente durante el tratamiento con ATRIPLA®. Debe realizarse la vigilancia sistemática de la depuración de creatinina calculada y del fósforo sérico en todos los pacientes con riesgo de disfunción renal, incluidos los pacientes que han sufrido anteriormente reacciones renales mientras recibían HEPSERA (dipivoxilo de adefovir). Se debe evitar el uso de ATRIPLA® con el uso reciente o concomitante de un fármaco nefrotóxico. Embarazo y reproducción: Embarazo de categoría D: El efavirenz puede causar daño fetal si se administra durante el primer trimestre del embarazo. En las mujeres que reciben tratamiento con ATRIPLA® debe evitarse el embarazo. Debe emplearse siempre un método anticonceptivo de barrera asociado a otros métodos anticonceptivos (por ejemplo, anticonceptivos orales u otros hormonales). Debido a la prolongada vida media del efavirenz, se recomienda el uso de medidas anticonceptivas durante 12 semanas después de la suspensión de ATRIPLA®. Las mujeres en edad fértil deberán someterse a una prueba de embarazo antes del inicio del tratamiento con ATRIPLA®. Si este fármaco se usa durante el primer trimestre del embarazo o si la paciente se queda embarazada mientras toma este fármaco, se le deberá informar acerca del posible daño para el feto. No hay estudios adecuados y bien controlados de ATRIPLA® en las mujeres embarazadas. ATRIPLA® deberá usarse durante el embarazo sólo si las posibles ventajas superan al posible riesgo para el feto, por ejemplo, en el caso de una embarazada sin otras opciones terapéuticas. Registro de embarazos con antirretrovíricos (Antiretoviral Pregnancy Registry): Se anima a los profesionales a cargo de la atención de los pacientes que registren a las pacientes que queden embarazadas en el siguiente sitio de Internet: www.kendle.com/registries/. Efavirenz: A fecha de julio de 2008, el Registro de embarazos con antirretrovíricos ha recibido informes prospectivos de 526 embarazos expuestos a pautas de tratamiento que contienen efavirenz; casi todos ellos se trataron de exposiciones durante el primer trimestre (507 embarazos). Se produjeron defectos congénitos en 13 de 407 nacidos vivos (exposición durante el primer trimestre) y dos de 37 nacidos vivos (exposición durante el segundo o tercer trimestre). Uno de estos defectos notificados prospectivamente con exposición durante el primer trimestre fue un defecto del tubo neural. Se ha notificado también prospectivamente un caso único de anoftalmía con la exposición durante el primer trimestre al efavirenz; sin embargo, este caso incluyó hendiduras faciales oblicuas y bandas amnióticas, una asociación conocida con la anoftalmía. Ha habido cinco notificaciones retrospectivas de descubrimientos compatibles con defectos del tubo neural, incluso meningomielocele. Todas las madres estuvieron expuestas a pautas que contenían efavirenz durante el primer trimestre. Aunque no se ha establecido una relación de causalidad entre estas reacciones y el empleo de efavirenz, se han observado defectos similares en estudios preclínicos de este fármaco. En un estudio de toxicidad del desarrollo se han observado malformaciones en tres de 20 fetos o lactantes de monos Cynomolgus tratados con efavirenz (en comparación con 0 de 20 testigos concomitantes). Las monas preñadas recibieron el fármaco durante toda la preñez (días 20 a 150 después del coito), con 60 mg/kg diarios de efavirenz, una dosis que produjo concentraciones plasmáticas del fármaco parecidas a las de los seres humanos que recibieron 600 mg/día de efavirenz. En un feto se observaron anencefalia y anoftalmía unilateral, con aumento de tamaño secundario de la lengua; en otro feto, microftalmia, y en un tercero, paladar hendido. En los monos Cynomolgus, el efavirenz atraviesa la placenta y produce concentraciones en la sangre fetal parecidas a las concentraciones en la sangre materna. Se ha comprobado que el efavirenz atraviesa la placenta en las ratas y los conejos, y produce concentraciones del fármaco en la sangre fetal parecidas a las concentraciones maternas. Se observó un aumento de las resorciones fetales en las ratas, con dosis de efavirenz que produjeron concentraciones máximas en el plasma y valores de AUC en las ratas hembras equivalentes o inferiores a las alcanzadas en seres humanos que recibieron 600 mg de efavirenz, una vez al día. El efavirenz no produjo ninguna toxicidad reproductiva cuando se administró a conejas preñadas, a dosis que produjeron concentraciones plasmáticas máximas parecidas y valores de AUC de aproximadamente la mitad a los alcanzados en seres humanos que recibieron 600 mg de efavirenz, una vez al día. Erupción cutánea: En ensayos clínicos controlados, el 26% (266/1008) de los pacientes tratados con 600 mg de efavirenz presentaron erupción cutánea de aparición reciente, en comparación con el 17% (111/635) de los pacientes tratados en los grupos testigos. La erupción asociada a flictenas, descamación húmeda o ulceración se presentó en el 0.9% (9/1008) de los pacientes tratados con efavirenz. La incidencia de erupción de grado 4 (por ejemplo, eritema multiforme, síndrome de Stevens-Johnson) en los pacientes tratados con efavirenz en todos los estudios y con acceso ampliado fue del 0.1%. Las erupciones cutáneas fueron generalmente maculopapulares, de gravedad leve a moderada, que se producen en las dos primeras semanas después del inicio del tratamiento con efavirenz (la mediana del tiempo hasta la aparición de la erupción en los adultos fue de 11 días) y, en la mayoría de los pacientes que continuaron el tratamiento con efavirenz, la erupción cutánea se resuelve al cabo de un mes (mediana de la duración, 16 días). En los ensayos clínicos, la tasa de suspensión a causa de una erupción cutánea fue del 1.7% (17/1008). La administración de ATRIPLA® puede reiniciarse en los pacientes que interrumpen el tratamiento a causa de una erupción. ATRIPLA® deberá suspenderse en los pacientes que presenten una erupción cutánea grave asociada a flictenas, descamación, afectación de las mucosas o fiebre. La administración de antihistamínicos o corticoesteroides adecuados puede mejorar la tolerabilidad y acelerar la resolución de la erupción. La experiencia con el efavirenz en pacientes que suspendieron la administración de otros antirretrovíricos del tipo de INNRT es limitada. Diecinueve pacientes que suspendieron la administración de nevirapina a causa de una erupción han sido tratados con efavirenz. Nueve de estos pacientes presentaron una erupción cutánea leve a moderada mientras recibían tratamiento con efavirenz, y dos de estos pacientes suspendieron el tratamiento a causa de una erupción. Enzimas hepáticas: Se recomienda la vigilancia de las enzimas hepáticas en los pacientes con antecedentes comprobados o presuntos de infección de hepatitis B o C, y en los pacientes tratados con otros medicamentos asociados a la toxicidad hepática. En los pacientes con aumentos persistentes de las transaminasas séricas superiores al quíntuple del límite superior de la normalidad, debe sopesarse la ventaja de continuar el tratamiento con ATRIPLA® frente a los riesgos desconocidos de una toxicidad hepática significativa [véase Reacciones adversas]. Disminución de la densidad mineral ósea: Se debe plantear la vigilancia de la densidad mineral ósea (DMO) en los pacientes infectados por el VIH-1 que tienen antecedentes de fracturas óseas patológicas o con riesgo de osteopenia. Si bien no se ha estudiado el efecto de los suplementos de calcio y vitamina D, dichos suplementos pueden ser beneficiosos para todos los pacientes. Se debe obtener asesoramiento adecuado si se sospecha de la presencia de anomalías óseas. En un estudio de 144 semanas en pacientes tratados con FD tenofovir sin tratamiento antirretrovírico previo, se observaron disminuciones en la densidad mineral ósea en la columna lumbar y la cadera, en ambos grupos de tratamiento del estudio. En la semana 144 hubo una disminución significativamente mayor del porcentaje medio con respecto al valor inicial de la DMO de la columna lumbar en los pacientes tratados con FD tenofovir + lamivudina + efavirenz, en comparación con los pacientes tratados con estavudina + lamivudina + efavirenz. Los cambios de la DMO en la cadera fueron similares entre los dos grupos de tratamiento. En ambos grupos, la mayor parte de la disminución de la DMO se produjo en las primeras 24 a 48 semanas del estudio y se mantuvo durante 144 semanas. El 28% de los pacientes tratados con FD tenofovir, frente al 21% de los pacientes del grupo de comparación, experimentaron una reducción de al menos el 5% en la DMO en la columna o del 7% en la DMO en la cadera. Se notificaron fracturas clínicamente importantes (sin incluir los dedos de los pies y las manos) en cuatro pacientes del grupo tratado con FD tenofovir y en seis pacientes del grupo de comparación. El FD tenofovir se asoció con aumentos significativos de los marcadores bioquímicos del metabolismo óseo (fosfatasa alcalina sérica específica del hueso, osteocalcina sérica, telopéptido C sérico y telopéptido N urinario), lo que sugiere un aumento del recambio de tejido óseo. Las concentraciones séricas de la hormona paratiroidea y de la vitamina D 1,25 también fueron más altas en los pacientes tratados con FD tenofovir. Se desconocen los efectos de los cambios asociados al FD tenofovir en la DMO y en los marcadores bioquímicos sobre la salud ósea a largo plazo y sobre el riesgo futuro de fracturas. Para obtener información adicional, consulte la ficha técnica del FD tenofovir. Se han notificado casos de osteomalacia (asociada a tubulopatía renal proximal y que puede contribuir a fracturas) en relación con el uso del FD tenofovir [véase Reacciones adversas]. Convulsiones: Se han observado convulsiones, en casos muy infrecuentes, en los pacientes tratados con efavirenz, generalmente en presencia de antecedentes comprobados de convulsiones. Debe tenerse precaución en cualquier paciente con antecedentes de convulsiones. Los pacientes que reciben concomitantemente medicamentos anticonvulsivos metabolizados principalmente por el hígado, como la fenitoína, la carbamacepina y el fenobarbital, pueden precisar la vigilancia periódica de las concentraciones plasmáticas [véase Interacciones]. Síndrome de reconstitución inmunológica: Se han notificado casos del síndrome de reconstitución inmunológica en los pacientes que recibieron tratamiento antirretrovírico asociado, incluidos los componentes de ATRIPLA®. Durante la fase inicial del tratamiento antirretrovírico asociado, los pacientes cuyo sistema inmunitario responde pueden presentar una respuesta inflamatoria ante infecciones oportunistas residuales o indolentes (por ejemplo, infección por Mycobacterium avium, citomegalovirus, neumonía por Pneumocystis jirovecii o tuberculosis), que pueden requerir evaluación y tratamiento adicionales. Redistribución de las grasas: Se ha observado la redistribución o la acumulación de la grasa corporal, consistente en obesidad central, acumulación de grasa dorsocervical ("joroba de búfalo"), emaciación periférica, emaciación facial, aumento de tamaño de las mamas y "aspecto cushingoide" en los pacientes en tratamiento antirretrovírico. Se desconocen actualmente el mecanismo y las consecuencias a largo plazo de estos fenómenos. No se ha establecido una relación causal.
Interacciones.
En este apartado se explican las interacciones farmacológicas clínicamente importantes observadas con ATRIPLA®. Los estudios de interacción farmacológica se describen en otro lugar de la ficha técnica [véase Farmacología]. Efavirenz: Se ha comprobado que el efavirenz induce in vivo el CYP3A. Otros compuestos que son sustratos del CYP3A pueden presentar una disminución de las concentraciones plasmáticas cuando se administran simultáneamente con efavirenz. En los estudios in vitro se ha demostrado que el efavirenz inhibe las isoenzimas CYP-2C9, 2C19 y 3A4, dentro de los límites de las concentraciones plasmáticas observadas de efavirenz. La administración concomitante de efavirenz con fármacos que son metabolizados principalmente por estas isoenzimas puede producir una alteración de las concentraciones plasmáticas del fármaco administrado concomitantemente. Por lo tanto, puede ser necesario ajustar adecuadamente las dosis de estos fármacos. Se esperaría que los fármacos que inducen la actividad del CYP3A4 (por ejemplo, fenobarbital, rifampicina, rifabutina) aumenten la depuración del efavirenz, produciéndose una disminución de las concentraciones plasmáticas. Emtricitabina y fumarato de disoproxilo de tenofovir: Como la emtricitabina y el tenofovir se eliminan principalmente por los riñones, la administración simultánea de ATRIPLA® con fármacos que reducen la función renal o que compiten por la secreción tubular activa puede aumentar las concentraciones séricas de la emtricitabina, el tenofovir o de otros fármacos eliminados por vía renal. Algunos ejemplos son, entre otros, el aciclovir, el dipivoxilo de adefovir, el cidofovir, el ganciclovir, el valaciclovir y el valganciclovir. La administración concomitante de FD tenofovir y didanosina debe realizarse con precaución, y se debe controlar estrictamente a los pacientes que reciben esta asociación, a fin de detectar reacciones adversas asociadas a la didanosina. La didanosina deberá suspenderse en los pacientes que presenten reacciones adversas relacionadas con este fármaco [ver en la tabla 7 las recomendaciones de ajuste de la posología de la didanosina]. Se ha observado la supresión de los recuentos de linfocitos CD4+ en los pacientes que recibieron FD tenofovir con 400 mg de didanosina diariamente. Se ha demostrado que la asociación de lopinavir y ritonavir aumenta las concentraciones de tenofovir. Se desconoce el mecanismo de esta interacción. Se debe vigilar la presencia de reacciones adversas asociadas al tenofovir en los pacientes que reciben la asociación de lopinavir y ritonavir con ATRIPLA®. La administración de ATRIPLA® deberá suspenderse en los pacientes que presenten reacciones adversas relacionadas con el tenofovir (ver la tabla 7). No se recomienda la administración concomitante de atazanavir con ATRIPLA®, ya que se ha comprobado que la administración concomitante de atazanavir con efavirenz o FD tenofovir disminuye las concentraciones plasmáticas de atazanavir. Asimismo, se ha demostrado que el atazanavir aumenta las concentraciones de tenofovir. No hay información suficiente para respaldar las recomendaciones de posología de atazanavir o de la asociación de atazanavir y ritonavir en asociación con ATRIPLA® (ver la tabla 7). Efavirenz, emtricitabina y fumarato de disoproxilo de tenofovir: En las tablas 6 y 7 se resume otra información importante acerca de las interacciones farmacológicas de ATRIPLA®. Las interacciones farmacológicas descritas se basan en estudios realizados con efavirenz, emtricitabina o FD tenofovir como fármacos individuales o son posibles interacciones farmacológicas. No se ha realizado ningún estudio de interacción farmacológica con ATRIPLA® (ver la información farmacocinética en Farmacología, tablas 1 a 5). En las tablas se incluyen las interacciones posiblemente significativas, pero no se incluyen todas.
Interferencia de efavirenz con los análisis: Interacción con los análisis de cannabinoides: El efavirenz no se une a los receptores de los cannabinoides. Se han observado resultados falsamente positivos de las pruebas de cannabinoides en la orina en voluntarios no infectados por el VIH que recibían efavirenz cuando se usó el análisis Cedia DAU Multi-Level THC de Microgenics para detección. Se obtuvieron resultados negativos cuando se realizó un análisis confirmatorio más específico con cromatografía de gases/espectrometría de masas. Para obtener más información, consúltese la ficha técnica de STOCRIN. Uso en poblaciones específicas: Embarazo: Categoría D del embarazo [ver Advertencias]. Lactancia: Los Centros para el Control y la Prevención de Enfermedades (de EE.UU.) recomiendan a las madres infectadas por el VIH-1 que no den lactancia materna a sus hijos a fin de evitar el riesgo de transmisión posnatal del VIH-1. En estudios en ratas se ha demostrado que tanto el efavirenz como el tenofovir se secretan por la leche. Se desconoce si el efavirenz, la emtricitabina o el tenofovir se excretan por la leche de los seres humanos. Debido a la posibilidad tanto de transmisión del VIH-1 como de reacciones adversas graves en los lactantes que reciben lactancia materna, se debe indicar a las madres que no den leche materna a los niños si están recibiendo ATRIPLA®. Empleo en pediatría: No se recomienda el uso de ATRIPLA® en los pacientes menores de 18 años, porque es un comprimido de asociación de dosis fijas que contiene un componente, el fumarato de disoproxilo de tenofovir, cuya inocuidad y eficacia no se han establecido en este grupo de edad. Empleo en geriatría: En los estudios clínicos de efavirenz, emtricitabina o FD tenofovir no se incluyó un número suficiente de pacientes con una edad a partir de 65 años como para determinar si responden de manera diferente a los pacientes más jóvenes. En general, la selección de la dosis para los pacientes ancianos debe ser cautelosa y se debe tener en cuenta la mayor frecuencia de disminución de las funciones cardiaca, renal o hepática, y las enfermedades concomitantes u otros tratamientos con fármacos. Disfunción renal: Dado que ATRIPLA® es una asociación de dosis fijas, no deberá recetarse a los pacientes que precisan un ajuste de la dosificación, por ejemplo, los que sufren una disfunción renal moderada o grave (depuración de creatinina < 50 ml/min) [ver Advertencias]. Disfunción hepática: No se han estudiado las características farmacocinéticas del efavirenz en los pacientes con disfunción hepática. Debido al extenso metabolismo del efavirenz mediado por el citocromo P450 y a la limitada experiencia clínica en los pacientes con disfunción hepática, debe tenerse precaución al administrar ATRIPLA® a estos pacientes [ver Advertencias]. Toxicología no clínica: Carcinogénesis, mutagénesis, trastorno de la fertilidad: Efavirenz: Se realizaron estudios de carcinogenia a largo plazo con efavirenz en ratones y ratas. Se administraron a los ratones dosis de 0, 25, 75, 150 ó 300 mg/kg al día, durante dos años. Las incidencias de adenomas y carcinomas hepatocelulares, y de adenomas alveolares o bronquiolares del pulmón aumentaron con respecto a los valores de referencia en las hembras. En los ratones machos no se observó ningún aumento de la incidencia de tumores con respecto a los valores de referencia. En los estudios en los que las ratas recibieron efavirenz, a dosis de 0, 25, 50 ó 100 mg/kg al día, durante dos años, no se observó ningún aumento de la incidencia de tumores con respecto a los valores de referencia. La exposición sistémica (basada en las AUC) en los ratones fue de aproximadamente 1.7 vez la de seres humanos que recibieron la dosis de 600 mg/día. La exposición en las ratas fue inferior a la de los seres humanos. Se desconoce el mecanismo del potencial carcinógeno. Sin embargo, en análisis de toxicología genética, el efavirenz no mostró indicios de actividad mutágena ni clastógena en un grupo de estudios in vitro e in vivo. Estos consistieron en análisis de mutación bacteriana en S. typhimurium y E. coli, análisis de mutación en mamíferos en las células de ovario de hámster chino, análisis de aberración cromosómica en los linfocitos humanos en la sangre periférica o en células de ovario de hámster chino, y análisis de micronúcleos en la médula ósea del ratón in vivo. Dada la ausencia de actividad genotóxica del efavirenz, se desconoce la pertinencia para los seres humanos de las neoplasias en los ratones tratados con efavirenz. El efavirenz no alteró el apareamiento ni la fertilidad de ratas machos o hembras, y no afectó a los espermatozoides de ratas machos. El rendimiento reproductivo de las crías nacidas de ratas que recibieron efavirenz no se vio afectado. A consecuencia de la rápida depuración del efavirenz en las ratas, las exposiciones sistémicas al fármaco alcanzadas en estos estudios fueron equivalentes o inferiores a las alcanzadas en los seres humanos que recibieron dosis terapéuticas de efavirenz. Emtricitabina: En estudios de carcinogenia a largo plazo con emtricitabina, no se observó ningún aumento de la incidencia tumoral relacionado con el fármaco en los ratones que recibieron dosis de hasta 750 mg/kg al día (26 veces la exposición sistémica en los seres humanos a la dosis terapéutica de 200 mg/día) ni en las ratas, con dosis de hasta 600 mg/día (31 veces la exposición sistémica en los seres humanos, a la dosis terapéutica). La emtricitabina no fue genotóxica en la prueba bacteriana de mutación inversa (prueba de Ames), el linfoma de ratón ni los análisis de micronúcleos de rata. La emtricitabina no afectó a la fertilidad en las ratas machos, con exposiciones (AUC) aproximadamente 140 veces superiores, ni en los ratones machos y hembras, con exposiciones aproximadamente 60 veces superiores a las exposiciones en los seres humanos, con la dosis recomendada de 200 mg diarios. La fertilidad fue normal en las crías de los ratones expuestos diariamente desde antes del nacimiento (exposición intrauterina) hasta la maduración sexual, a exposiciones diarias (AUC) aproximadamente 60 veces superiores a las exposiciones en los seres humanos, con la dosis recomendada de 200 mg diarios. Fumarato de disoproxilo de tenofovir: Se realizaron estudios de carcinogenia oral a largo plazo con FD tenofovir en ratones y ratas, a exposiciones de hasta aproximadamente 16 veces (ratones) y cinco veces (ratas) las observadas en los seres humanos con la dosis terapéutica para la infección por el VIH-1. Con la dosis alta en los ratones hembras, aumentaron los adenomas hepáticos con exposiciones de hasta 16 veces superiores a la de los seres humanos. En las ratas, el estudio arrojó resultados negativos en cuanto a la carcinogenia, con exposiciones de hasta cinco veces las observadas en los seres humanos con la dosis terapéutica. El fumarato de disoproxilo de tenofovir fue mutágeno en el análisis in vitro de linfoma de ratón, y negativo en la prueba de mutagénesis bacteriana in vitro (prueba de Ames). En un análisis de micronúcleos de ratón in vivo, el FD tenofovir fue negativo cuando se administró a ratones machos. No se produjeron efectos sobre la fertilidad, el rendimiento del apareamiento ni el desarrollo embrionario temprano al administrar FD tenofovir a ratas machos, a una dosis equivalente a diez veces la dosis en los seres humanos, a partir de comparaciones de la superficie corporal, durante 28 días antes del apareamiento, y a ratas hembras, durante 15 días antes del apareamiento, hasta el séptimo día de gestación. Sin embargo, en las ratas hembras hubo una alteración del ciclo menstrual. Toxicología o farmacología en animales: Efavirenz: Se observaron convulsiones no sostenidas en seis de veinte monos que recibieron efavirenz a dosis que produjeron valores plasmáticos del AUC de 4 a 13 veces superiores a los de los seres humanos que recibieron la dosis recomendada. Fumarato de disoproxilo de tenofovir: El tenofovir y el fumarato de disoproxilo de tenofovir, administrados a ratas, perros y monos, en estudios toxicológicos con exposiciones (basadas en las AUC) superiores o iguales a 6 veces las observadas en los seres humanos, ocasionaron toxicidad ósea. En los monos, la toxicidad ósea se diagnosticó como osteomalacia. La osteomalacia observada en los monos parecía ser reversible al reducir la dosis o suspender el tenofovir. En las ratas y los perros, la toxicidad ósea se manifestó en forma de disminución de la densidad del mineral óseo. Se desconoce el mecanismo o mecanismos subyacentes de la toxicidad ósea. En cuatro especies de animales a las que se administró tenofovir y FD tenofovir se observaron pruebas de toxicidad renal. En estos animales se observaron aumentos en la creatinina sérica, BUN (nitrógeno ureico en sangre), glucosuria, proteinuria, fosfaturia o calciuria, y disminución del fosfato sérico en diferentes grados. Estas toxicidades se observaron en exposiciones (según las AUC) de 2 a 20 veces superiores a las observadas en los seres humanos. Se desconoce la relación de las anomalías renales, en especial de la fosfaturia, con la toxicidad ósea. Estudios clínicos: El estudio clínico 934 respalda el uso de los comprimidos de ATRIPLA® en los pacientes infectados por el VIH-1 sin tratamiento previo con antirretrovíricos. Puede encontrarse información adicional en respaldo del uso de ATRIPLA® en los pacientes sin tratamiento previo con antirretrovíricos en la ficha técnica de VIREAD. El estudio clínico 073 respalda el uso de ATRIPLA® en los pacientes con tratamiento previo con antirretrovíricos. En este estudio se presenta la experiencia clínica del uso de ATRIPLA® en pacientes estables y virológicamente suprimidos, con tratamiento antirretrovírico asociado, sin antecedentes de fracaso virológico. En los pacientes con tratamiento previo con antirretrovíricos, el uso de los comprimidos de ATRIPLA® también puede plantearse en los pacientes con cepas del VIH-1 que se espera que sean sensibles a los componentes de ATRIPLA®, según la evaluación de la historia o mediante pruebas genotípicas o fenotípicas [Ver Farmacología]. Estudio 934: Se informan los datos obtenidos en 144 semanas en el estudio 934, un estudio multicéntrico, aleatorizado, abierto y con testigo activo, en que se comparó emtricitabina + FD tenofovir, administrados en asociación con efavirenz, frente a la asociación en dosis fijas de zidovudina y lamivudina, en asociación con efavirenz, en 511 pacientes sin tratamiento previo con antirretrovíricos. De