BARACLUDE®
BMS
Análogo nucleósido inhibidor de la transcriptasa reversa activo frente al virus de la hepatitis B.
Descripción.
BARACLUDE® es el nombre comercial de entecavir, un nucleósido análogo de guanosina con actividad selectiva contra el VHB. El nombre químico para entecavir es monohidrato de 2-amino-1,9-dihidro-9-{(1S, 3R, 4S)-4-hidroxi-3-(hidroximetil)-2-metilenciclopentil}-6H-purin-6-ona. Su fórmula molecular es C12H15N5O3·H2O, la cual corresponde a un peso molecular de 295,3. Entecavir tiene la siguiente fórmula estructural: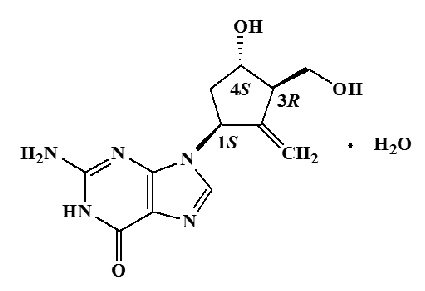
Entecavir es un polvo de color blanco a blanquecino. Es ligeramente soluble en agua (2,4mg/mL), y el pH de la solución saturada en agua es 7,9 a 25°C ±0,5.
Composición.
Comprimidos recubiertos: cada comprimido recubierto contiene: entecavir (como monohidrato) 0,5mg, 1mg. Excipientes: lactosa monohidrato, Celulosa microcristalina, Crospovidona, Povidona, Estearato de magnesio, Opadry blanco YS-1-18027-A (dióxido de titanio, hipromelosa, polietilenglicol 400, polisorbato 80), Opadry rosado, 03B14899 (dióxido de titanio, hipromelosa, polietilenglicol 400, óxido de hierro rojo).
Farmacología.
Mecanismo de acción: entecavir es un fármaco antiviral. Farmacocinética: se evaluó la farmacocinética de dosis sencilla y múltiple de entecavir en sujetos saludables y en sujetos con infección crónica por el virus de la hepatitis B. Absorción: después de la administración oral en sujetos saludables, las concentraciones plasmáticas máximas de entecavir se alcanzaron entre las 0,5 y 1,5 horas. Después de la administración de múltiples dosis diarias entre 0,1 y 1,0 mg, la Cmáx y el área bajo la curva (ABC) concentración-tiempo en estado de equilibrio se incrementaron proporcionalmente con la dosis. El estado de equilibrio se alcanzó después de 6-10 días de administrar una dosis única diaria con una acumulación aproximada de 2 veces. Para una dosis oral de 0,5 mg, la Cmáx en estado de equilibrio fue de 4,2 ng/ml y la concentración mínima en plasma (Cmín) fue de 0,3 ng/ml. Para una dosis oral de 1 mg, la Cmáx fue de 8,2 ng/ml y la Cmín fue de 0,5 ng/ml. En sujetos saludables, la biodisponibilidad de la tableta/comprimido fue del 100%, comparada con la solución oral. La solución oral y la tableta/comprimido pueden ser utilizados indistintamente. Efecto de los alimentos en la absorción oral: la administración oral de 0,5mg de entecavir con una comida estándar alta en grasa (945 kcal, 54,6 g de grasa) o una comida liviana (379 kcal, 8,2 g de grasa) produjo una demora en la absorción (de 1,0-1,5 horas en presencia de alimento versus 0,75 horas en ayuno), una reducción en la Cmáx de 44-46%, y una disminución en el ABC de 18-20% [ver Dosificación]. Distribución: con base en el perfil farmacocinético de entecavir después de una dosis oral, el volumen aparente de distribución estimado supera al agua corporal total, lo cual indica que entecavir se distribuye extensamente en los tejidos. La unión de entecavir a proteínas del suero humano in vitro fue del 13% aproximadamente. Metabolismo y eliminación: después de la administración de 14C-entecavir en humanos y ratas, no se observaron metabolitos oxidativos o acetilados. Se observaron cantidades menores de metabolitos de fase 2 (glucurónidos y sulfatos conjugados). Entecavir no es un sustrato, inhibidor o inductor, del sistema enzimático citocromo P450 (CYP450) [ver Interacciones]. Después de alcanzar la concentración máxima, las concentraciones de entecavir en plasma disminuyeron de una manera biexponencial con un tiempo de vida media de eliminación terminal de aproximadamente 128-149 horas. El índice de acumulación observada del fármaco es aproximadamente de 2 veces con una dosis diaria, lo que sugiere una vida media de acumulación eficaz de 24 horas aproximadamente. Entecavir se elimina predominantemente por el riñón, con una recuperación urinaria del fármaco sin cambios en el estado de equilibrio que varía entre 62 y 73% de la dosis administrada. El aclaramiento renal es independiente de la dosis y varía entre 360 y 471 mL/min lo que sugiere que entecavir experimenta tanto filtración glomerular como secreción tubular [ver Interacciones]. Poblaciones especiales: género: no hay diferencias significativas entre los géneros en relación con la farmacocinética de entecavir. Raza: no hay diferencias significativas entre las razas en relación con la farmacocinética de entecavir. Edad avanzada: el efecto de la edad sobre la farmacocinética de entecavir fue evaluado después de la administración de una única dosis oral de 1 mg en voluntarios sanos jóvenes y en voluntarios de edad avanzada. El ABC de entecavir fue un 29,3% mayor en los sujetos de edad avanzada en comparación con los sujetos jóvenes. La disparidad en la exposición entre los sujetos de edad avanzada y los jóvenes probablemente sea atribuible a diferencias en la función renal. El ajuste de la dosis de BARACLUDE® deberá basarse en la función renal del paciente, en lugar de la edad [ver Dosificación]. Pediatría: no se han realizado estudios farmacocinéticos en niños. Deterioro renal: la farmacocinética de entecavir después de una dosis única de 1 mg fue estudiada en sujetos (sin infección crónica por el virus de la hepatitis B) con grados seleccionados de deterioro renal, incluyendo sujetos cuyo deterioro renal fue controlado por hemodiálisis o diálisis peritoneal ambulatoria continua (DPAC). Los resultados se muestran en la Tabla 5 [ver Dosificación].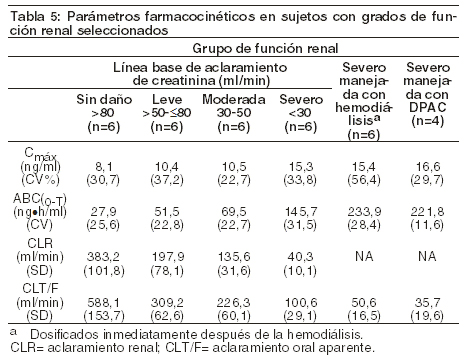
Después de una dosis única de 1 mg de entecavir administrado 2 horas antes de la sesión de hemodiálisis, la hemodiálisis removió aproximadamente el 13% de la dosis en 4 horas. La DPAC removió aproximadamente 0,3% de la dosis en 7 días [ver Dosificación]. Deterioro hepático: la farmacocinética de entecavir tras una dosis única de 1 mg fue estudiada en sujetos (sin infección crónica por el virus de la hepatitis B) con deterioro hepático moderado o severo (Child-Pugh clases B o C). La farmacocinética de entecavir fue similar en sujetos con deterioro hepático y en sujetos sanos control; por tanto, no se recomienda un ajuste en la dosis de BARACLUDE® para pacientes con deterioro hepático. Postrasplante hepático: la seguridad y la eficacia de BARACLUDE® en receptores de trasplante hepático aún se desconocen. No obstante, en un estudio piloto pequeño sobre el uso de entecavir en receptores de trasplante de hígado infectados con VHB que recibían una dosis estable de ciclosporina A (n=5) o tacrolimus (n=4), la exposición a entecavir fue aproximadamente 2 veces superior a la exposición en voluntarios sanos con función renal normal. La función renal alterada contribuyó al incremento en la exposición a entecavir en estos sujetos. Las interacciones farmacocinéticas potenciales entre entecavir y ciclosporina A o tacrolimus no han sido formalmente evaluadas [ver Uso en poblaciones específicas]. Interacciones medicamentosas: el metabolismo de entecavir fue evaluado en estudios in vivo e in vitro. Entecavir no es un sustrato, inhibidor o inductor del sistema enzimático citocromo P450 (CYP450). A concentraciones hasta 10,000 veces mayores que aquellas obtenidas en humanos, entecavir no inhibió ninguna de las enzimas principales del CYP450 humano 1A2, 2C9, 2C19, 2D6, 3A4, 2B6 y 2E1. A concentraciones hasta 340 veces mayores de las observadas en humanos, entecavir no indujo las enzimas 1A2, 2C9, 2C19, 3A4, 3A5 y 2B6 del CYP450 humano. Es poco probable que la farmacocinética de entecavir se afecte por la coadministración con agentes que son metabolizados por, que inhiben o que inducen el sistema CYP450. De la misma manera, es poco probable que la farmacocinética de los sustratos conocidos de CYP se vea afectada por la coadministración de entecavir. La farmacocinética en estado de equilibrio de entecavir y del fármaco coadministrado no se vio alterada en estudios de interacción de entecavir con lamivudina, adefovir dipivoxil y tenofovir disoproxil fumarato [ver Interacciones]. Microbiología: mecanismo de acción: entecavir, un análogo nucleósido de la guanosina que posee actividad contra la polimerasa del VHB, se fosforila eficazmente en la forma activa como trifosfato, con una vida media intracelular de 15 horas. Al competir con el sustrato natural desoxiguanosina trifosfato, el trifosfato de entecavir inhibe funcionalmente las tres actividades de la polimerasa del VHB (transcriptasa reversa, rt): (1) cebado de la base, (2) transcripción reversa de la porción negativa del ARN mensajero pregenómico y (3) síntesis de la porción positiva del ADN del VHB. El trifosfato de entecavir es un inhibidor débil de las polimerasas a, b y d del ADN celular y de la polimerasa c del ADN mitocondrial, con valores de Ki que oscilan de 18 a > 160 mM. Actividad antiviral: entecavir inhibió la síntesis del ADN del VHB (reducción del 50%, EC50) a una concentración de 0,004M en células humanas HepG2 transfectadas con el VHB del tipo salvaje. La mediana del valor de EC50 para entecavir contra el VHB resistente a la lamivudina (rtL180M, rtM204V) fue de 0,026 mM (rango entre 0,010-0,059 mM). Es poco probable que la coadministración de inhibidores nucleósidos/nucleótidos de la transcriptasa reversa del VIH (NRTIs) con BARACLUDE® reduzca la eficacia antiviral de BARACLUDE® contra el VHB o de alguno de estos agentes contra el VIH. En cultivos celulares, las combinaciones del VHB con abacavir, didanosina, lamivudina, estavudina, tenofovir, o zidovudina no presentaron efecto antagonista contra la actividad anti-VHB de entecavir dentro de un amplio rango de concentraciones. En ensayos antivirales contra el VIH, entecavir no presentó efecto antagonista sobre la actividad anti-VIH en cultivo celular de estos seis NRTIs o emtricitabina a concentraciones mayores que 100 veces la Cmáx de entecavir utilizando la dosis de 1 mg. Actividad antiviral frente al VIH: un análisis completo de la actividad inhibitoria de entecavir frente a un panel de aislados, de laboratorio y clínicos, del VIH tipo 1 (VIH-1) utilizando una variedad de células y condiciones de ensayo arrojó valores de la EC50 entre 0,026 a > 10 mM; los valores de la EC50 más bajos se observaron cuando en el ensayo se utilizaron niveles decrecientes del virus. En cultivo celular, entecavir seleccionó una sustitución en la posición M184I en la transcriptasa inversa del VIH, en concentraciones micromolares, confirmando la presión inhibitoria a concentraciones altas de entecavir. Las variantes del VIH que contenían la sustitución en la posición M184V mostraron pérdida de susceptibilidad al entecavir. Resistencia: en cultivos celulares: en ensayos en células, se observaron reducciones de 8 a 30 veces en la susceptibilidad fenotípica a entecavir en las cepas resistentes a lamivudina. Posteriores reducciones ( > 70 veces) en la susceptibilidad fenotípica a entecavir requirieron la presencia de sustituciones de aminoácidos (rtM204I/V con o sin rtL180M) junto con sustituciones adicionales en los residuos rtT184, rtS202 o rtM250, o una combinación de estas sustituciones con o sin una sustitución en la posición rtI169 de la polimerasa del VHB. Resistencia cruzada: se ha observado resistencia cruzada entre los análogos nucleósidos del VHB. En ensayos celulares, entecavir tuvo de 8 a 30 veces menos inhibición de la síntesis de ADN del VHB para el VHB que contiene sustituciones de resistencia a la lamivudina y la telbivudina rtM204I/V con o sin rtL180M que del tipo salvaje de VHB. Las sustituciones rtM204I/V con o sin rtL180M, rtL80I/V o rtV173L, que están asociadas a la resistencia a la lamivudina y la telbivudina, también confieren susceptibilidad fenotípica disminuida al entecavir. En los estudios clínicos, no se estableció la eficacia de entecavir contra el VHB que contiene sustituciones asociadas a la resistencia a adefovir. Las cepas aisladas del VHB provenientes de sujetos con resistencia a lamivudina que no toleraron la terapia con entecavir se mantuvieron susceptibles a adefovir en cultivos celulares, pero mantuvieron la resistencia a la lamivudina. Los genomas recombinantes del VHB que codifican sustituciones asociadas a resistencia a adefovir, en la posición rtN236T o rtA181V presentaron cambios de 0,3 y 1,1 veces mayores en la susceptibilidad a entecavir en cultivos celulares, respectivamente. Toxicología no clínica: carcinogénesis, mutagénesis, deterioro de la fertilidad: estudios de carcinogenicidad oral a largo plazo con entecavir fueron llevados a cabo a exposiciones 42 veces (ratones) y 35 veces (ratas) más altas que las observadas en humanos a la dosis más alta recomendada de 1 mg/día. En los estudios en ratas y ratones, entecavir fue positivo para hallazgos carcinogénicos. En ratones, se incrementaron los adenomas de pulmón en hembras y machos a exposiciones 3 y 40 veces mayores que la exposición en humanos. Los carcinomas de pulmón se incrementaron tanto en ratones machos como hembras con exposiciones 40 veces mayores que la exposición en humanos. Los adenomas combinados con carcinomas pulmonares aumentaron en ratones machos con exposiciones 3 veces mayores y en hembras con exposiciones 40 veces mayores que la exposición en humanos. El desarrollo del tumor estuvo precedido por proliferación de neumocitos en el pulmón, lo cual no se observó en ratas, perros o monos que recibieron entecavir, en apoyo a la conclusión de que los tumores de pulmón en ratón pueden ser un evento específico de la especie. Los carcinomas hepatocelulares se incrementaron en machos y los adenomas hepáticos combinados con carcinomas también se incrementaron con exposiciones 42 veces mayores que la exposición en humanos. Tumores vasculares en ratones hembra (hemangiomas de ovario y útero y hemangiosarcomas de bazo) se incrementaron a exposiciones 40 veces mayores que la exposición en humanos. En ratas, se incrementaron los adenomas hepatocelulares en hembras con exposiciones 24 veces mayores que la exposición en humanos; adenomas y carcinomas combinados también se incrementaron en hembras con exposiciones 24 veces mayores que la exposición en humanos. Gliomas cerebrales fueron inducidos tanto en hembras como en machos con exposiciones 35 y 24 veces mayores que la exposición en humanos. Fibromas cutáneos fueron inducidos en hembras con exposiciones 4 veces mayores que la exposición en humanos. Se desconoce en qué medida los resultados de estudios de carcinogenicidad en roedores pueden ser predictivos para los humanos. Entecavir fue clastogénico en cultivos de linfocitos humanos. Entecavir no fue mutagénico en el ensayo de mutación inversa bacteriana de Ames usando cepas de S. typhimurium y E. coli en presencia o ausencia de activación metabólica, en un ensayo de mutación genética de células de mamíferos y en un ensayo de transformación con células embrionales de hámster sirio. Entecavir también fue negativo en un estudio oral de micronúcleo y en un estudio oral de reparación de ADN en ratas. En estudios de toxicología reproductiva, en los cuales los animales recibieron una dosis hasta de 30 mg/kg de entecavir por un período de hasta cuatro semanas, no se observó evidencia de daño en la fertilidad en ratas hembra ni macho a exposiciones sistémicas > 90 veces que aquellas alcanzadas en humanos a la dosis más alta recomendada de 1 mg/día. En estudios toxicológicos en roedores y perros, se observó degeneración de los tubos seminíferos a exposiciones ≥35 veces que aquellas alcanzadas en humanos. No se observaron cambios testiculares en monos. Resultados más allá de las 48 semanas: la duración óptima del tratamiento con BARACLUDE® se desconoce. De acuerdo con los criterios estipulados en el protocolo de los estudios clínicos fase 3, los sujetos discontinuaron su tratamiento con BARACLUDE® o lamivudina después de 52 semanas de acuerdo con una definición de respuesta basada en la supresión virológica del VHB ( < 0,7MEq/ml mediante el ensayo de ADNb) y pérdida de HBeAg (en sujetos HBeAg-positivos) o ALT < 1,25 x LSN (en sujetos HBeAg-negativos) en la semana 48. Los sujetos que alcanzaron la supresión virológica pero no tuvieron respuesta serológica (HBeAg-positivos) o no alcanzaron un nivel de ALT < 1,25 x LSN (HBeAg-negativos) continuaron recibiendo tratamiento ciego durante 96 semanas o hasta alcanzar los criterios de respuesta. Estas pautas para el manejo de sujetos especificadas en el protocolo no están destinadas a servir de guía en la práctica clínica. Sujetos sin tratamiento previo con nucleósidos: entre los sujetos HBeAg-positivos sin tratamiento previo con nucleósidos (Estudio AI463022), 243 (69%) sujetos tratados con BARACLUDE® y 164 (46%) sujetos tratados con lamivudina continuaron recibiendo tratamiento ciego por un período de hasta 96 semanas. Entre quienes continuaron el tratamiento a ciego en el año 2, 180 (74%) sujetos tratados con BARACLUDE® y 60 (37%) sujetos tratados con lamivudina alcanzaron un nivel de ADN del VHB < 300 copias/ml según PCR realizada al finalizar el tratamiento (hasta 96 semanas). Ciento noventa y tres (79%) sujetos tratados con BARACLUDE® alcanzaron un nivel de ALT ≤1 x LSN en comparación con 112 (68%) sujetos tratados con lamivudina, y la seroconversión del HBeAg ocurrió en 26 (11%) sujetos tratados con BARACLUDE® y en 20 (12%) sujetos tratados con lamivudina. Entre los sujetos HBeAg-positivos sin tratamiento previo con nucleósidos, 74 (21%) sujetos tratados con BARACLUDE® y 67 (19%) sujetos tratados con lamivudina alcanzaron la definición de respuesta en la semana 48, dejaron de recibir los fármacos del estudio y se les hizo un seguimiento fuera del tratamiento durante 24 semanas. Entre quienes respondieron al tratamiento con BARACLUDE®, 26 (35%) sujetos tenían un nivel de ADN del VHB < 300 copias/ml, 55 (74%) sujetos habían alcanzado un nivel de ALT ≤1 x LSN y 56 (76%) sujetos mantuvieron la seroconversión del HBeAg al finalizar el seguimiento. Entre quienes respondieron a lamivudina, 20 (30%) sujetos tenían un nivel de ADN del VHB < 300 copias/ml, 41 (61%) sujetos habían alcanzado un nivel de ALT ≤1 x LSN y 47 (70%) sujetos mantuvieron la seroconversión del HBeAg al finalizar el seguimiento. Entre los sujetos HBeAg-negativos sin tratamiento previo con nucleósidos (Estudio AI463027), 26 (8%) sujetos tratados con BARACLUDE® y 28 (9%) sujetos tratados con lamivudina continuaron con el tratamiento ciego por un período de hasta 96 semanas. En esta cohorte pequeña que continuó el tratamiento en el año 2, 22 sujetos tratados con BARACLUDE® y 16 tratados con lamivudina tuvieron un nivel de ADN del VHB < 300 copias/ml por PCR, y 7 y 6 sujetos, respectivamente, habían alcanzado un nivel de ALT ≤1 x LSN al finalizar el tratamiento (hasta un período de 96 semanas). Entre los sujetos HBeAg-negativos sin tratamiento previo con nucleósidos, 275 (85%) sujetos tratados con BARACLUDE® y 245 (78%) sujetos tratados con lamivudina alcanzaron la definición de respuesta en la semana 48, suspendieron los fármacos del estudio y se les hizo un seguimiento fuera del tratamiento durante 24 semanas. En esta cohorte, muy pocos sujetos en cada grupo de tratamiento tuvieron un nivel de ADN del VHB < 300 copias/ml por PCR al finalizar el seguimiento. Al finalizar el seguimiento, 126 (46%) sujetos tratados con BARACLUDE® y 84 (34%) sujetos tratados con lamivudina habían alcanzado un nivel de ALT ≤1 x LSN. Sujetos con resistencia a lamivudina: entre los sujetos con resistencia a lamivudina (Estudio AI463026), 77 (55%) sujetos tratados con BARACLUDE® y 3 (2%) sujetos tratados con lamivudina continuaron el tratamiento ciego por un período de hasta 96 semanas. En esta cohorte de sujetos bajo tratamiento con BARACLUDE®, 31 (40%) sujetos alcanzaron un nivel de ADN del VHB < 300 copias/ml, 62 (81%) sujetos habían alcanzado un nivel de ALT ≤1 x LSN y 8 (10%) sujetos demostraron seroconversión del HBeAg al finalizar el tratamiento. Poblaciones especiales: pacientes coinfectados con VIH y VHB: AI463038 fue un estudio randomizado, doble-ciego, controlado con placebo con BARACLUDE® versus placebo en 68 sujetos coinfectados con VIH y VHB y que experimentaron recurrencia de viremia por VHB mientras recibían un régimen de terapia antirretroviral altamente activa (HAART) con lamivudina. Los sujetos continuaron su régimen HAART con lamivudina (dosis de lamivudina de 300 mg/día) y se les adicionó bien fuera BARACLUDE® de 1 mg una vez al día (51 sujetos) o un placebo (17 sujetos) por 24 semanas seguido por una fase de etiqueta abierta por 24 semanas adicionales donde todos los sujetos recibieron BARACLUDE®. En la línea de base, los sujetos tuvieron una media del nivel de ADN del VHB en suero por PCR de 9,13 log10 copias/ml. El 99% de los sujetos fueron HBeAg-positivos en la línea de base, con un nivel de ALT medio de 71,5U/L. El nivel medio de ARN del VIH se mantuvo estable en aproximadamente 2 log10 copias/ml durante las 24 semanas de tratamiento ciego. Los criterios de valoración bioquímicos y virológicos a la Semana 24 se muestran en la Tabla 10. No hay datos en pacientes con coinfección por VIH/VHB que no habían recibido tratamiento previo con lamivudina. BARACLUDE® no fue evaluado en pacientes coinfectados con VIH/VHB que no estaban recibiendo simultáneamente un tratamiento eficaz para el VIH [ver Advertencias y Precauciones].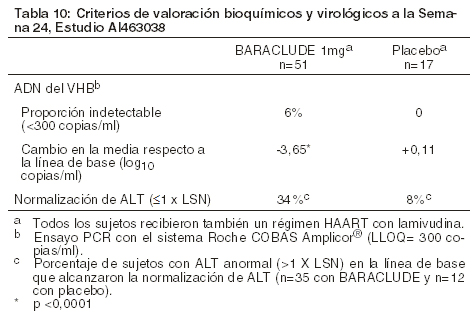
En los sujetos que fueron asignados inicialmente a BARACLUDE®, al finalizar la fase de tratamiento abierto (semana 48), el 8% de los sujetos tenían un nivel de ADN del VHB < 300 copias/ml por PCR, el cambio en la media con respecto a la línea de base del nivel de ADN del VHB por PCR fue de -4,20 log10 copias/ml, y el 37% de los sujetos con un nivel anormal de ALT en la línea de base había alcanzado la normalización de ALT (≤1 x LSN).
Indicaciones.
BARACLUDE® está indicado para el tratamiento de la infección crónica causada por el virus de la hepatitis B en adultos con evidencia de replicación viral activa o con evidencia de elevaciones persistentes en las aminotransferasas séricas (ALT o AST) o enfermedad histológicamente activa. Los siguientes puntos deben considerarse al iniciar el tratamiento con BARACLUDE®: esta indicación se basa en las respuestas histológicas, virológicas, bioquímicas y serológicas en sujetos adultos sin tratamiento previo con nucleósidos y resistentes a lamivudina con infección crónica por VHB HBeAg positivo o HBeAg negativo con enfermedad hepática compensada [ver Estudios clínicos]. Están disponibles datos virológicos, bioquímicos y serológicos de un estudio controlado en sujetos adultos con infección crónica por VHB y enfermedad hepática descompensada [ver Reacciones Adversas y Estudios clínicos]. Están disponibles datos virológicos, bioquímicos y serológicos para un número limitado de sujetos adultos con coinfección por VIH/VHB que recibieron previamente tratamiento con lamivudina [ver Advertencias y Precauciones].
Dosificación.
Posología/Dosis y Administración: BARACLUDE® debe ser administrado con el estómago vacío (al menos 2 horas antes y 2 horas después de comer). Dosis recomendada: Enfermedad Hepática Compensada: La dosis recomendada de BARACLUDE para infección crónica por virus de hepatitis B en adultos y adolescentes mayores de 16 años sin tratamiento previo con nucleósidos es de 0,5 mg una vez al día. La dosis recomendada de BARACLUDE® en adultos y adolescentes de por lo menos 16 años de edad con historia de viremia por hepatitis B mientras reciben lamivudina o con mutaciones conocidas de resistencia a lamivudina o telbivudina en la posición rtM204I/V con o sin las posiciones rtL180M, rtL80I/V o rtV173L es de 1 mg una vez al día. Enfermedad Hepática Descompensada: La dosis recomendada de BARACLUDE® para infección crónica por virus de hepatitis B en adultos con enfermedad hepática descompensada es de 1 mg una vez al día. Deterioro renal: en sujetos con deterioro renal, el aclaramiento oral aparente de entecavir disminuyó cuando disminuyó el aclaramiento de creatinina [ver Farmacología]. Se recomienda ajustar la dosis para pacientes con aclaramiento de creatinina < 50ml/min, incluyendo pacientes en hemodiálisis o diálisis peritoneal ambulatoria continua (DPAC), como se muestra en la Tabla 1. Se prefieren los regímenes de dosis única diaria.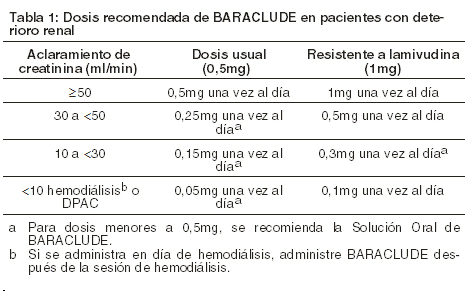
Deterioro hepático: no es necesario ajustar la dosis para pacientes con deterioro hepático. Duración del tratamiento: la duración óptima del tratamiento con BARACLUDE® para pacientes con infección crónica causada por el virus de la hepatitis B y la relación entre tratamiento y resultados a largo plazo como cirrosis y carcinoma hepatocelular son aún desconocidas. Formas de dosificación y concentraciones: BARACLUDE® 0,5mg en comprimidos recubiertos blancos a grisáceos, de forma triangular, con "BMS" moldeado a un lado y "1611" en el otro lado. BARACLUDE® 1mg en comprimidos recubiertos rosados, de forma triangular, con "BMS" moldeado a un lado y "1612" en el otro lado.
Contraindicaciones.
BARACLUDE® está contraindicado en pacientes con hipersensibilidad previamente demostrada a entecavir o a cualquier componente del producto.
Reacciones adversas.
Las siguientes reacciones adversas se analizan con mayor detalle en otras secciones del prospecto: exacerbaciones de la hepatitis después de discontinuar el tratamiento [ver recuadro de Advertencias]. Acidosis láctica y hepatomegalia severa con esteatosis [ver recuadro de Advertencia]. Experiencia de los estudios clínicos: dado que los estudios clínicos se llevan a cabo en condiciones muy diversas, los índices de reacciones adversas observados en los estudios clínicos de un fármaco no pueden compararse directamente con los índices de los estudios clínicos de otro fármaco y es posible que no reflejen los índices observados en la práctica. La evaluación de las reacciones adversas está basada en cuatro estudios (AI463014, AI463022, AI463026 y AI463027) en los cuales 1720 sujetos con infección crónica por el virus de la hepatitis B recibieron tratamiento doble-ciego con BARACLUDE® 0,5mg/día (n=679), BARACLUDE® 1 mg/día (n=183) o lamivudina (n=858) hasta por 2 años. La mediana de duración de la terapia fue de 69 semanas para los sujetos tratados con BARACLUDE® y de 63 semanas para los sujetos tratados con lamivudina en los Estudios AI463022 y AI463027, y de 73 semanas para los sujetos tratados con BARACLUDE® y de 51 semanas para los sujetos tratados con lamivudina en los Estudios AI463026 y AI463014. Los perfiles de seguridad de BARACLUDE® y lamivudina fueron comparables en estos estudios. Las reacciones adversas más comunes de cualquier severidad (3%) con al menos una relación posible con el fármaco de estudio para sujetos tratados con BARACLUDE® fueron dolor de cabeza, fatiga, mareos y náuseas. Las reacciones adversas más comunes entre los sujetos tratados con lamivudina fueron dolor de cabeza, fatiga y mareos. El uno por ciento de los sujetos tratados con BARACLUDE® en estos cuatro estudios comparado con el 4% de los sujetos tratados con lamivudina discontinuaron el tratamiento por eventos adversos o por resultados de pruebas de laboratorio anormales. Reacciones adversas clínicas de intensidad entre moderada a severa y consideradas al menos como posiblemente relacionadas con el tratamiento en cuatro estudios clínicos en los cuales se comparó a BARACLUDE® con lamivudina son presentadas en la Tabla 2.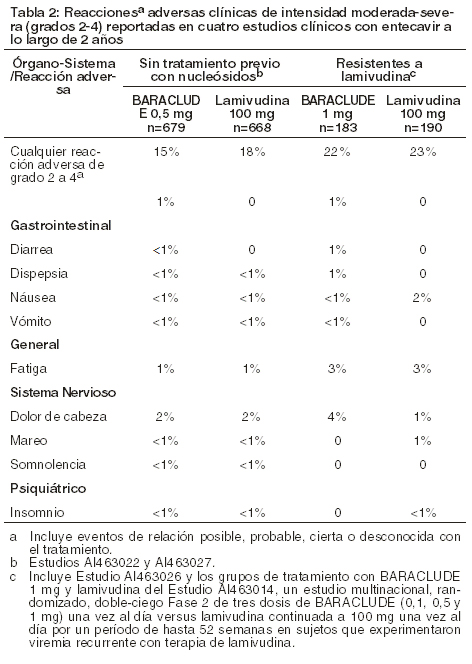
Anormalidades de laboratorio: en la Tabla 3 se detalla la frecuencia de anormalidades de laboratorio seleccionadas, emergentes del tratamiento y reportadas durante la terapia en cuatro estudios clínicos en los cuales se comparó a BARACLUDE® con lamivudina.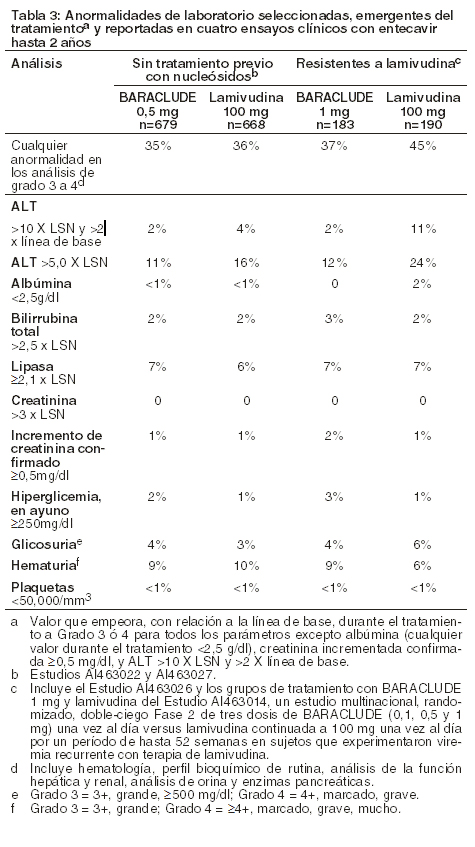
Entre los sujetos tratados con BARACLUDE® en estos estudios, las elevaciones de ALT durante el tratamiento mayores que 10 veces el límite superior de la normalidad (LSN) y mayores que 2 veces la línea de base generalmente se resolvieron con la continuación del tratamiento. La mayoría de estas exacerbaciones estuvo asociada con una reducción ≥2 log10 en la carga viral precedida o coincidente con la elevación de ALT. Se recomienda monitorización periódica de la función hepática durante el tratamiento. Exacerbaciones de la hepatitis después de discontinuar el tratamiento: La exacerbación de la hepatitis o el aumento de la ALT se definió como un nivel ALT > 10 x LSN y > 2 x el nivel de referencia del sujeto (medición mínima en la línea de base o última medición al finalizar la dosis). La Tabla 4 presenta la proporción de sujetos en cada estudio que experimentaron aumentos de ALT posteriores al tratamiento de todos los sujetos que discontinuaron el tratamiento (independientemente del motivo). En estos estudios, a un subgrupo de sujetos se les permitió que discontinuaran el tratamiento en la semana 52 o posteriormente si habían alcanzado la respuesta al tratamiento definida por el protocolo. Si se discontinúa BARACLUDE® sin tener en cuenta la respuesta al tratamiento, el índice de exacerbaciones post-tratamiento podría ser mayor.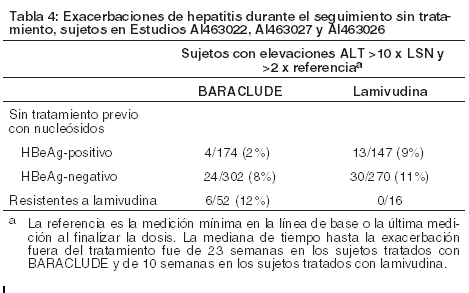
Enfermedad Hepática Descompensada: El AI463048 fue un estudio randomizado, abierto, de BARACLUDE® 1 mg una vez al día en comparación con adefovir dipivoxil 10 mg una vez al día administrados, durante un máximo de 48 semanas, a sujetos adultos con infección crónica por el VHB y evidencia de descompensación hepática, definida con un puntaje de Child-Turcotte-Pugh (CTP) de 7 o mayor [ver Estudios Clínicos]. Entre los 102 sujetos tratados con BARACLUDE®, los eventos adversos emergentes del tratamiento más frecuentes y de cualquier gravedad, independientemente de la causalidad, que se produjeron hasta la Semana 48, fueron edema periférico (16%), ascitis (15%), pirexia (14%), encefalopatía hepática (10%), e infección respiratoria alta (10%). Las reacciones adversas clínicas no enumeradas en la Tabla 2 y observadas hasta la Semana 48 incluyen disminución del bicarbonato en sangre (2%) e insuficiencia renal ( < 1%). Dieciocho de los 102 (18%) sujetos tratados con BARACLUDE® y 18 de los 89 (20%) sujetos tratados con adefovir dipivoxil murieron durante las primeras 48 semanas de tratamiento. La mayoría de las muertes (11 en el grupo que recibió BARACLUDE® y 16 en el grupo que recibió adefovir dipivoxil) se debieron a causas hepáticas, como insuficiencia hepática, encefalopatía hepática, síndrome hepatorrenal y hemorragia digestiva alta. El índice de carcinoma hepatocelular (CHC) hasta la Semana 48 fue del 6% (6/102) en los sujetos que recibieron tratamiento con BARACLUDE® y del 8% (7/89) en los sujetos que recibieron tratamiento con adefovir dipivoxil. El 5% de los sujetos en ambos grupos de tratamiento interrumpió el tratamiento debido a un evento adverso en la Semana 48. En ambos grupos de tratamiento, ninguno de los sujetos presentó exacerbación hepática durante el tratamiento (ALT > 2 X nivel basal y > 10 X LSN) hasta la Semana 48. Once de 102 (11%) de los sujetos que recibieron tratamiento con BARACLUDE® y 11/89 (13%) de los sujetos tratados con adefovir dipivoxil tuvieron un aumento confirmado en la creatinina sérica de 0,5 mg/dl hasta la Semana 48. Coinfección por el VHB/VIH: El perfil de seguridad de BARACLUDE® 1 mg (n=51) administrado en sujetos coinfectados por el VHB/VIH enrolados en el estudio AI463038 fue similar al del placebo (n=17) durante 24 semanas de tratamiento ciego, y fue similar al registrado en los sujetos sin infección por el VIH [ver Advertencias y Precauciones]. Experiencia postcomercialización: las siguientes reacciones adversas han sido reportadas durante el uso posaprobación de BARACLUDE®. Debido a que estas reacciones fueron reportadas voluntariamente a partir de una población de tamaño desconocido, no siempre es posible estimar en forma confiable su frecuencia o establecer una relación causal con la exposición a BARACLUDE®. Trastornos del sistema inmunológico: reacción anafiláctica. Trastornos metabólicos y nutricionales: Acidosis Láctica. Trastornos hepatobiliares: Aumento de las transaminasas. Trastornos en la piel y el tejido subcutáneo: alopecia, erupción.
Advertencias.
Advertencias y Precauciones:
Exacerbaciones agudas severas de la hepatitis B: exacerbaciones agudas severas de la hepatitis B han sido reportadas en pacientes que han discontinuado el tratamiento antihepatitis B, incluyendo entecavir [ver Reacciones adversas)]. La función hepática debe ser estrechamente monitorizada con seguimiento clínico y de laboratorio al menos por unos meses después de discontinuar el tratamiento antihepatitis B. Si es apropiado, la reanudación del tratamiento antihepatitis B puede ser garantizada. Pacientes coinfectados con VIH y VHB: BARACLUDE® no ha sido evaluado en pacientes coinfectados con VIH/VHB que no estaban tratados simultáneamente con un tratamiento efectivo frente al VIH. La experiencia clínica limitada sugiere que existe una posibilidad para el desarrollo de la resistencia del VIH a los nucleósidos inhibidores de la transcriptasa reversa si BARACLUDE® es utilizado para tratar la infección crónica por el virus de la hepatitis B en pacientes con infección por el VIH que no están siendo tratados [ver Farmacología clínica]. Por lo tanto, el tratamiento con BARACLUDE® no está recomendado en los pacientes coinfectados por VIH/VHB que no estén recibiendo al mismo tiempo una terapia antirretroviral altamente activa (HAART). Antes de iniciar el tratamiento con BARACLUDE®, se ofrecerá a todos los pacientes realizar el análisis de anticuerpos frente al VIH. BARACLUDE® no ha sido estudiado como tratamiento de la infección por VIH y no está recomendado para este uso. Acidosis láctica y hepatomegalia severa con esteatosis: acidosis láctica y hepatomegalia severa con esteatosis, incluyendo casos fatales, han sido reportadas con el uso de análogos nucleósidos solos o en combinación con antirretrovirales. La mayoría de estos casos se han reportado en mujeres. La obesidad y la prolongada exposición a los nucleósidos pueden ser factores de riesgo. Se debe tener especial precaución cuando se administren análogos de nucleósidos a pacientes con factores conocidos de riesgo para enfermedad hepática; sin embargo, también se han reportado casos en pacientes sin factores de riesgo conocidos. Se han informado casos de acidosis láctica con el uso de BARACLUDE, a menudo asociados con descompensación hepática, otras afecciones médicas graves o exposiciones a fármacos. Es posible que los pacientes con enfermedad hepática descompensada tengan un mayor riesgo de sufrir acidosis láctica. El tratamiento con BARACLUDE® debe suspenderse en cualquier paciente que desarrolle problemas clínicos o de laboratorio que sugieran acidosis láctica o hepatotoxicidad pronunciada (que puede incluir hepatomegalia y esteatosis incluso en ausencia de una marcada elevación de las transaminasas).
Interacciones.
Como el entecavir es principalmente eliminado por los riñones [ver Farmacología clínica], la coadministración de BARACLUDE® con fármacos que reducen la función renal o que compiten por la secreción tubular activa puede aumentar las concentraciones séricas tanto de entecavir como del fármaco coadministrado. La coadministración de entecavir con lamivudina, adefovir dipivoxil o tenofovir disoproxil fumarato no resultó en interacciones significativas de fármacos. Los efectos de la coadministración de BARACLUDE® con otros fármacos que se eliminan por vía renal o que se conoce que afectan la función renal no han sido evaluados y los pacientes deben ser monitorizados de cerca por eventos adversos cuando BARACLUDE® es coadministrado con tales fármacos. Uso en poblaciones específicas: embarazo: embarazo categoría C: no hay estudios adecuados y bien controlados de BARACLUDE® en mujeres embarazadas. Cuando se administró entecavir a ratas y conejos gestantes a exposiciones 28 y 212 veces la exposición humana a la máxima dosis humana, no hubo signos de toxicidad embriofetal. Debido a que los estudios de reproducción en animales no son siempre predictivos de la respuesta en humanos, BARACLUDE® debe ser usado durante el embarazo solo si es evidentemente necesario y después de cuidadosas consideraciones de los riesgos y beneficios. Se realizaron estudios de desarrollo de toxicidad en ratas y conejos. No hubo signos de toxicidad embriofetal o materna cuando se les administró a las ratas y los conejos gestantes dosis orales de entecavir a exposiciones de aproximadamente 28 (rata) y 212 (conejo) veces la exposición humana alcanzada con la máxima dosis humana recomendada de 1mg/día. En ratas, se observó toxicidad materna, toxicidad embriofetal (resorciones), menor peso fetal, malformaciones de cola y vértebras, osificación reducida (vértebras, esternebras y falanges) y vértebras lumbares y costillas adicionales se observaron a exposiciones 3100 veces superiores a la exposición humana. En conejos se observó toxicidad embriofetal (resorciones), osificación reducida (hioide) y una creciente incidencia de la 13° costilla con exposiciones 883 veces superiores a la humana. En un estudio perinatal y postnatal, no se evidenciaron efectos adversos en la descendencia cuando se administró entecavir oralmente a ratas a exposiciones > 94 veces la dosis en humanos. Trabajo de parto y parto: no hay estudios en mujeres embarazadas, como tampoco hay datos sobre el efecto de BARACLUDE® en la transmisión del VHB de la madre al infante. Por tanto, se deben utilizar intervenciones apropiadas para prevenir el contagio neonatal con VHB. Madres lactantes: no se conoce si BARACLUDE® es excretado en la leche humana; sin embargo, entecavir es excretado en la leche de ratas. Debido a que muchos fármacos se excretan en la leche humana y debido al potencial de reacciones adversas graves en los lactantes por BARACLUDE®, se debe adoptar una decisión de suspender la lactancia o suspender BARACLUDE® tomando en consideración la importancia de la continuación del tratamiento contra la hepatitis B en la