BENLYSTA
GLAXOSMITHKLINE
Tratamiento del herpes eritematoso sistémico.
Composición.
Polvo liofilizado estéril en un vial monodosis. Vial de 120 mg: Cada vial contiene una cantidad suficiente de Belimumab para proporcionar 120 mg en 1,5 mL al llevar a cabo la reconstitución de acuerdo a las recomendaciones con agua inyectable estéril. Después de la reconstitución, cada mL de solución contiene 80 mg de Belimumab. Excipientes: Ácido cítrico monohidrato, Citrato de sodio dihidrato, Sucrosa, Polisorbato 80. Belimumab es un anticuerpo monoclonal IgG1l recombinante completamente humano. Presentación farmacéutica: Polvo para solución para perfusión.
Farmacología.
Farmacodinamia: Mecanismo de acción: El estimulador de Linfocitos B (BLyS, también llamado BAFF y TNFSF13), un miembro de la familia de ligandos del factor de necrosis tumoral (TNF), que inhibe la apoptosis de células B y estimula la diferenciación de células B a células plasmáticas productoras de inmunoglobulinas. El BLyS se sobre expresa en pacientes con LES. Existe una fuerte asociación entre la actividad del LES (evaluada mediante el Índice de Actividad de la Enfermedad Lupus Eritematoso Sistémico-Seguridad de los Estrógenos en la Evaluación Nacional de Lupus Eritematoso [SELENA-SLEDAI]) y las concentraciones plasmáticas de BLyS. Belimumab es un anticuerpo monoclonal IgG1l completamente humano que se fija específicamente al BLyS humano soluble e inhibe su actividad biológica. Belimumab no se fija directamente a las células B, pero al unirse al BLyS, Belimumab inhibe la supervivencia de las células B, incluyendo a las células B autor reactivas, y reduce la diferenciación de células B a células plasmáticas productoras de inmunoglobulinas. Efecto farmacodinámico: Se observaron reducciones en las concentraciones séricas elevadas de IgG y en los anticuerpos anti-DNA de doble cadena desde la Semana 8, y continuaron hasta la Semana 52. En pacientes con hipergammaglobulinemia en la línea basal, se observó una normalización de las concentraciones de IgG en la Semana 52 en 49% y 20% de los pacientes que recibieron Belimumab y placebo, respectivamente. En pacientes con anticuerpos anti-DNA de doble cadena en la línea basal, las reducciones en pacientes que estaban recibiendo Belimumab fueron evidentes desde la Semana 8, y para la Semana 52, 16% de los pacientes tratados con Belimumab se habían convertido a anti-DNA de doble cadena negativos, en comparación con 7% de los pacientes tratados con placebo. En pacientes con niveles bajos de complemento en la línea basal, el tratamiento con Belimumab produjo incrementos en el complemento que se observaron desde la Semana 4 y se mantuvieron a través del tiempo. Para la Semana 52, las concentraciones de C3 y C4 se habían normalizado en 38% y 44% de los pacientes que estaban recibiendo Belimumab, en comparación con 17% y 19% de los pacientes tratados con placebo. El blanco de Belimumab, BLyS, es una citoquina crucial para la supervivencia, diferenciación y proliferación de las células B. Belimumab redujo significativamente las células B circulantes, vírgenes, activadas y plasmáticas, así como el subconjunto de células B en pacientes con LES en la Semana 52. Las reducciones en las células vírgenes, plasmáticas y plasmáticas de vida corta, así como en el subconjunto de células B en pacientes con LES, se observaron desde la Semana 8. Las células de memoria aumentaron inicialmente y disminuyeron lentamente hacia los niveles basales en la Semana 52. En una extensión no controlada a largo plazo del estudio, se hizo seguimiento durante más de 7 años de tratamiento continuo de la células B (incluyendo células no activadas, activadas, células plasmáticas y el subtipo SLE de las células B) y de los niveles de IgG. Se observó una sustancial y sostenida disminución en varios subtipos de células B significando una disminución mediana de 87% en células B no activadas, 67% en células B de memoria, 99% en células B activadas, y 92% en células plasmáticas después de más de 7 años de tratamiento. Después de cerca de 7 años, se observó una reducción mediana de 28% en los niveles de IgG con 1.6% de los sujetos experimentando una disminución en los niveles de IgG por debajo de 400 mg/dL. Durante el curso del estudio, la incidencia reportada de AEs generalmente permaneció estable o declinó. Inmunogenicidad: El análisis de sensibilidad para la medicación de anticuerpos neutralizantes y anticuerpo no específico anti-fármaco (ADA) fue limitado dada la presencia del principio activo en las muestras recogidas. Se desconoce por tanto, la verdadera presencia de la neutralización de anticuerpos y de anticuerpo no específico anti-fármaco. En los dos estudios en Fase III, 4 de 563 (0,7%) pacientes en el grupo de 10 mg/kg, y 27 de 559 (4,8%) pacientes en el grupo de 1 mg/kg, desarrollaron anticuerpos anti-Belimumab persistentes. La frecuencia reportada para el grupo de 10mg/kg puede subestimar la frecuencia real debido a una menor sensibilidad del ensayo en presencia de altas concentraciones de droga. Anticuerpos neutralizantes fueron detectados en tres pacientes que recibidan belimumab 1mg/kg. Sin embargo, la presencia de anticuerpos anti-belimumab fue relativamente poco frecuente y no se puede obtener conclusiones en relación al efecto de la inmunogenicidad sobre la farmacocinética de belimumab debido al bajo número de sujetos positivos para anticuerpos anti-belimumab. En los estudios fase III, de los sujetos con anticuerpos anti-belimumab persistentemente positivos, 1/10 (10%), 2/27 (7%) y 1/4 (25%) en el grupo placebo, 1 mg/Kg y 10 mg/Kg, respectivamente, experimentaron reacciones relacionadas con la perfusión en el día de dosificación; estas reacciones relacionadas con la perfusión fueron de gravedad leve a moderada. En algunos pacientes con ADA se notificaron EAs graves/importantes. Las tasas de relaciones relacionadas con la perfusión entre los sujetos con anticuerpos anti-belimumab persistentemente positivos fue comparable a las tasas de pacientes ADA negativos 75/552 (14%), 78/523 (15%), y 83/559 (15%) en el grupo placebo, 1 mg/Kg y 10 mg/Kg, respectivamente. Farmacocinética: Los parámetros farmacocinéticos que se describen a continuación se basan en estimaciones paramétricas poblacionales que son específicas a los 563 pacientes que recibieron 10 mg/kg de Belimumab en los dos estudios en Fase III. Absorción: Belimumab se administra mediante perfusión intravenosa. Se observaron concentraciones séricas máximas de Belimumab al final de la perfusión, o poco después de ésta. La concentración sérica máxima fue de 313 mg/mL, con base en la simulación del perfil de concentración-tiempo utilizando los valores paramétricos típicos del modelo farmacocinético poblacional. Distribución: Belimumab se distribuyó en los tejidos con un volumen de distribución global de 5,29 L. Metabolismo: Belimumab es una proteína cuya vía metabólica esperada es la degradación a péptidos pequeños y aminoácidos individuales mediante enzimas proteolíticas ampliamente distribuidas. No se han realizado estudios de biotransformación clásica. Eliminación: Las concentraciones séricas de Belimumab disminuyeron de manera biexponencial, con una vida media de distribución de 1,75 días y una vida media terminal de 19,4 días. La depuración sistémica fue de 215 mL/día. Interacciones farmacológicas: El uso concomitante de micofenolato mofetilo, azatioprina e hidroxicloroquina no influyó sustancialmente en la farmacocinética de Belimumab, con base en los resultados del análisis farmacocinético poblacional. Un amplio rango de otros co-medicamentos (antiinflamatorios no esteroidales, aspirina, e inhibidores de la HMG-CoA reductasa) tampoco influyeron significativamente en la farmacocinética de Belimumab. La coadministración de esteroides e inhibidores de la ACE ocasionó un incremento estadísticamente significativo en la depuración sistémica en el análisis farmacocinético poblacional. Sin embargo, estos efectos no fueron clínicamente significativos, ya que su magnitud se mantuvo dentro del rango de variabilidad normal de la depuración. Grupos de pacientes especiales: Pacientes de edad avanzada: Belimumab ha sido estudiado en un número limitado de pacientes de edad avanzada. Dentro de la población global del estudio con LES IV, la edad no afectó la exposición a Belimumab en el análisis farmacocinético poblacional. Sin embargo, debido al pequeño número de sujetos de 65 años de edad o mayores, no se puede descartar definitivamente un efecto de la edad. Niños y adolescentes: No hay datos farmacocinéticos disponibles en pacientes pediátricos. Insuficiencia renal: No se han realizado estudios formales para examinar los efectos de la presencia de insuficiencia renal en la farmacocinética de Belimumab. Durante el desarrollo clínico, Belimumab se estudió en un número limitado de pacientes con LES con insuficiencia renal (depuración de creatinina < 60 mL/min, incluyendo un número pequeño con depuración de creatinina < 30 mL/min). Aunque la presencia de proteinuria ( > 2 g/día) aumentó la depuración de Belimumab, y las disminuciones en la depuración de creatinina disminuyeron la depuración de Belimumab, estos efectos se mantuvieron dentro del rango esperado de variabilidad. Por lo tanto, no se recomienda ajustar la dosis en pacientes con insuficiencia renal. Insuficiencia hepática: No se han realizado estudios formales para examinar los efectos de la presencia de insuficiencia hepática en la farmacocinética de Belimumab. Las moléculas IgG1 como Belimumab se catabolizan mediante enzimas proteolíticas ampliamente distribuidas, las cuales no están restringidas al tejido hepático; por lo tanto, no es probable que los cambios en la función hepática tengan algún efecto en la eliminación de Belimumab. Otras características de los pacientes: No se observó que el género, la raza o la etnicidad ejercieran un efecto significativo en la farmacocinética de Belimumab. Los efectos que ejerce el tamaño corporal en la exposición a Belimumab se encuentran justificados por la dosificación normalizada respecto al peso. Estudios clínicos: Se evaluó la eficacia de BENLYSTA en dos estudios aleatorizados, doble ciegos, controlados con placebo y en Fase III, realizados en 1.684 pacientes con diagnóstico clínico de LES de acuerdo con los criterios de clasificación del Colegio Americano de Reumatología. Los pacientes elegibles tenían LES activo, definido como una calificación de SELENA-SLEDAI mayor o igual a 6 y con resultados positivos en las pruebas (título de ANA ≥1:80 y/o positivos a anti-DNA de doble cadena [≥30 unidades/mL]) de anticuerpos anti-nucleares (ANA o anti-DNA de doble cadena) en la selección. Los pacientes se encontraban en un régimen terapéutico estable para LES (cuidado estándar) que consistía en cualquiera de los siguientes (solos o en combinación): corticoesteroides, anti-maláricos, AINEs u otros inmunosupresores. Los pacientes fueron excluidos del estudio si alguna vez habían recibido tratamiento con cualquier tratamiento dirigido a las células B, si habían recibido otro agente biológico en investigación dentro del año anterior, o si habían tenido una respuesta positiva a las pruebas de anticuerpos anti-HIV, de antígeno superficial de hepatitis B o de anticuerpos anti-hepatitis C. Los dos estudios tuvieron diseños similares, exceptuando que el Estudio 1 fue un estudio de 76 semanas y el Estudio 2 fue un estudio de 52 semanas. Ambos estudios tuvieron criterios primarios de valoración de 52 semanas. El Estudio 1 (HGS1006-C1056) se realizó principalmente en Norteamérica y Europa Occidental. La distribución racial fue 70% blancos/caucásicos, 14% negros/afroamericanos, 13% nativos de Alaska o indoamericanos y 3% asiáticos. Los medicamentos de base incluyeron corticoesteroides (76%), inmunosupresores (56%) y anti-maláricos (63%). El Estudio 2 (HGS1006-C1057) se realizó en Sudamérica, Europa del Este, Asia y Australia. La distribución racial fue 38% asiáticos, 26% blancos/caucásicos, 32% nativos de Alaska o indoamericanos y 4% negros/afroamericano. Los medicamentos de base incluyeron corticoesteroides (96%), inmunosupresores (42%) y anti-maláricos (67%). La mediana de edad de los pacientes en ambos estudios fue de 37 años (rango: 18 a 73 años), y la mayoría (94%) eran mujeres. En la selección, los pacientes fueron estratificados según la severidad de su enfermedad, con base en su calificación de SELENA-SLEDAI ( < 9 vs ≥10), nivel de proteinuria ( < 2 g por 24 hrs vs ≥2 g por 24 hrs) y raza, y luego fueron asignados aleatoriamente para recibir 1 mg/kg de BENLYSTA, 10 mg/kg de BENLYSTA o placebo, además del cuidado estándar. Se administró a los pacientes el medicamento del estudio por vía intravenosa durante un período de 1 hora los días 0, 14, 28, y luego cada 28 días durante 48 o 72 semanas. El criterio primario de valoración de eficacia fue un criterio de valoración compuesto (Índice de respondedor de LES) que definió la respuesta como el cumplimiento de cada uno de los siguientes criterios en la Semana 52, en comparación con la línea basal: - reducción de ≥4 puntos en la calificación de SELENA-SLEDAI, y - sin nueva calificación de dominio orgánico de acuerdo al G British Isles Lupus Assessment Group (BILAG A), o dos nuevos puntajes dominio orgánico de acuerdo al BILAG B, y - sin empeoramiento (incremento menor de 0,30 puntos) en la calificación de la Evaluación Global del médico (EGM). El índice de respondedor de LES utiliza la calificación de SELENA-SLEDAI como una medida objetiva de la reducción en la actividad global de la enfermedad; el índice de BILAG se utiliza para garantizar que no haya un agravamiento significativo en algún sistema orgánico específico; y la EGM se utiliza para garantizar que las mejorías en la actividad de la enfermedad no se deban a la condición global del paciente. BENLYSTA produjo mejorías significativas en el Índice de respondedor de LES, así como en la calificación del componente individual de SELENA-SLEDAI en ambos estudios; véase tabla.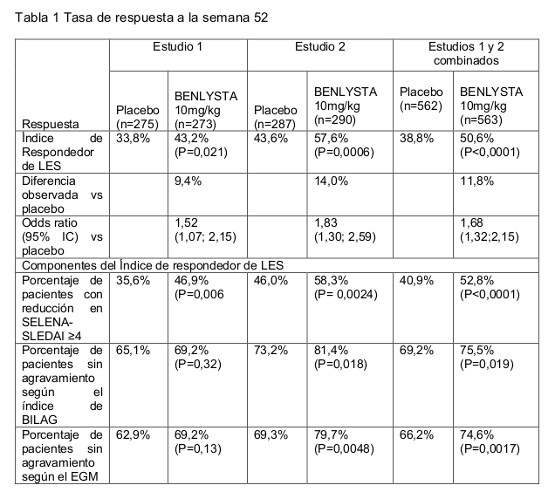
En un análisis combinado de los dos estudios, el porcentaje de pacientes que estaban recibiendo más de 7,5 mg/día de prednisona (o su equivalente) en la línea basal, y cuya dosis promedio de corticoesteroide se redujo por lo menos en 25% respecto a la línea basal a una dosis equivalente a menor o igual 7,5 mg/día de prednisona durante las Semanas 40 a 52, fue de 17,9% en el grupo que estaba recibiendo Belimumab y de 12,3% en el grupo tratado con placebo (P=0,0451). Los brotes de LES se definieron mediante el Índice Modificado de Brotes de LES de SELENA SLEDAI, en el que la modificación excluye brotes graves desencadenados únicamente por un incremento en la calificación de SELENA SLEDAI a un valor mayor que 12. La mediana de tiempo hasta el primer brote experimentó un retraso en el grupo combinado que estaba recibiendo Belimumab, en comparación con el grupo tratado con placebo (cociente de riesgo = 0,84, P=0,012). Los brotes graves se observaron en el 15,6% del grupo que recibió Benlysta frente al 23,7% del grupo placebo durante las 52 semanas de observación (diferencia de tratamiento observada = -8,1%; hazard ratio=0,64; p=0,0011). El análisis univariado y multivariado de los puntos finales primarios demostró que el mayor beneficio fue observado en pacientes con alta actividad de la enfermedad al inicio, incluyendo pacientes con escalas SELENA SLEDAI mayores o iguales a 10, pacientes que requieren esteroides para controlar su enfermedad y pacientes con bajos niveles de complemento. Un análisis Post-hoc identificó una alta respuesta en el subgrupo de pacientes con bajo nivel de complemento y anticuerpos anti dsDNA positivos al inicio, ver tabla 2. De estos pacientes el 64,5% tenía escala de SELENA SLEDAI mayor o igual a 10 con respecto al inicio.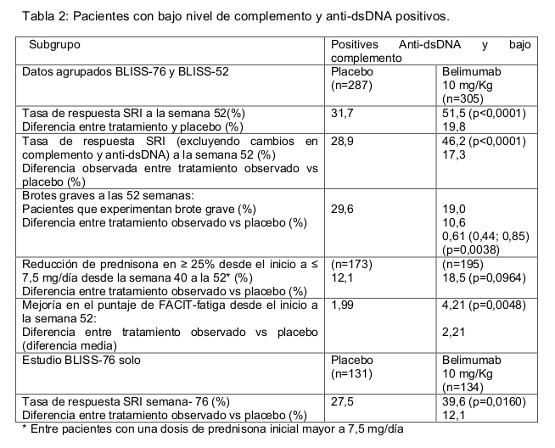
Hubo muy pocos pacientes varones, mayores de 65 años de edad, o negros/afroamericanos reclutados en los estudios clínicos controlados como para poder sacar conclusiones significativas acerca de los efectos que ejercen el género, la edad o la raza en los resultados clínicos. Datos preclínicos de seguridad: Los datos no clínicos no revelaron algún peligro especial para los humanos con base en los estudios de toxicidad con dosis repetidas y de toxicidad en la reproducción. La administración intravenosa y subcutánea en monos ocasionó una reducción esperada en el número de recuentos de células B periféricas y en tejido linfoide, sin observarse hallazgos toxicológicos asociados. Se han realizado estudios reproductivos en monos macacos preñados que estaban recibiendo 150 mg/kg de Belimumab mediante perfusión intravenosa (aproximadamente 9 veces la exposición clínica máxima anticipada en humanos) cada 2 semanas hasta por 21 semanas, y el tratamiento con Belimumab no se asoció con efectos perjudiciales directos o indirectos respecto a toxicidad materna, toxicidad en el desarrollo o teratogenicidad. Los hallazgos relacionados con el tratamiento se limitaron a una reducción reversible y esperada en los recuentos de células B, tanto en hembras como en lactantes, así como reducción reversible en los niveles de IgM en monos lactantes. Los números de células B volvieron a la normalidad después de suspender el tratamiento con Belimumab aproximadamente un año después del parto en monos adultos, y a los tres meses de vida en monos lactantes; los niveles de IgM en lactantes expuestos a Belimumab in útero volvieron a la normalidad a los seis meses de vida. Debido a que Belimumab es un anticuerpo monoclonal, no se han realizado estudios de genotoxicidad. No se han realizado estudios de carcinogenicidad o de fertilidad (machos o hembras).
Indicaciones.
BENLYSTA está indicado para reducir la actividad de la enfermedad en pacientes adultos con lupus eritematoso sistémico (LES) activo, positivo a autoanticuerpos, que estén recibiendo tratamiento estándar.
Dosificación.
Debe considerarse la discontinuación del tratamiento con BENLYSTA si no hay mejoría en el control de la enfermedad después de 6 meses de tratamiento. BENLYSTA se administra por vía intravenosa mediante perfusión, y debe reconstituirse y diluirse antes de la administración (ver Uso y Manejo). BENLYSTA debe ser administrado por un profesional entrenado en el tratamiento de reacciones de hipersensibilidad incluyendo la anafilaxia. BENLYSTA debe administrarse en perfusión durante un período de 1 hora. BENLYSTA no debe administrarse como "push" o bolo intravenoso. La velocidad puede disminuirse o interrumpirse si el paciente desarrolla una reacción a la perfusión. La perfusión debe suspenderse inmediatamente si el paciente experimenta una reacción adversa potencialmente mortal (ver Contraindicaciones, Advertencias). Se debe monitorear a los pacientes durante y por un período apropiado después de la administración de BENLYSTA (ver Advertencias, Reacciones Adversas). Premedicación en pacientes con alergias: Puede administrarse premedicación con un antihistamínico oral, con o sin un antipirético, antes de la perfusión de BENLYSTA (ver Advertencias, Estudios clínicos). Adultos: El régimen de dosificación recomendado es de 10 mg/kg los días 0, 14 y 28, y posteriormente en intervalos de 4 semanas. La interrupción del tratamiento con BENLYSTA debe valorarse si no existe mejoría en el control de la enfermedad tras 6 meses de tratamiento. Niños: BENLYSTA no ha sido estudiado en pacientes menores de 18 años de edad. No existen datos acerca de la seguridad y eficacia de BENLYSTA en este grupo de edad. Pacientes de edad avanzada: No se ha establecido la eficacia y seguridad de Benlysta en pacientes de edad avanzada. Los datos en pacientes mayores a 65 años, se limitan a menos del 1,6% de la población de estudio. Por lo tanto no se recomienda el uso de Benlysta en pacientes de edad avanzada a menos que los beneficios esperados superen los riesgos. En caso de que la administración de Benlysta a pacientes de edad avanzada se considere necesaria, no es necesario ajustar la dosis. Aunque los datos son limitados, no se recomienda ajuste de dosis (ver Farmacocinética - Poblaciones de pacientes especiales). Insuficiencia renal: No se han realizado estudios formales de BENLYSTA en pacientes con insuficiencia renal. BENLYSTA ha sido estudiado en un número limitado de pacientes con LES con insuficiencia renal. No se requiere ajustar la dosis en pacientes con insuficiencia renal (ver Farmacocinética - Poblaciones de pacientes especiales). Insuficiencia hepática: No se han realizado estudios formales de BENLYSTA en pacientes con insuficiencia hepática. Sin embargo, es poco probable que los pacientes con insuficiencia hepática requieran modificaciones de la dosis (ver Farmacocinética - Poblaciones de pacientes especiales).
Contraindicaciones.
BENLYSTA está contraindicado en pacientes que han demostrado anafilaxia a Belimumab o a alguno de los excipientes.
Reacciones adversas.
Se ha evaluado la seguridad de BENLYSTA en pacientes con LES en 3 estudios controlados con placebo y administración intravenosa, y un estudio controlado con placebo y administración subcutánea. Los datos descritos a continuación reflejan la exposición a BENLYSTA (10 mg/kg por vía intravenosa durante un período de 1 hora los días 0, 14, 28, y luego cada 28 días durante 52 semanas) en 674 pacientes con LES, incluyendo 472 expuestos hasta por 52 semanas y 556 pacientes expuestos a 200 mg de belimumab subcutáneo una vez a la semana durante 52 semanas. La información de seguridad presentada incluye datos posteriores a la semana 52 de algunos pacientes. También se incluye información de los reportes postmercadeo. La mayoría de los pacientes también recibieron uno o más de los siguientes tratamientos concomitantes para el LES: corticoesteroides, agentes inmunomoduladores, anti-maláricos, antiinflamatorios no esteroidales. En el 93% de los pacientes tratados con Benlysta y en el 92% de los pacientes tratados con placebo se notificaron reacciones adversas. Las reacciones adversas notificadas más frecuentemente (≥10% de los pacientes con LES tratados con Benlysta junto con el tratamiento estándar y en una tasa ≥1% mayor que placebo) fueron náusea, diarrea y pirexia. La proporción de pacientes que suspendió el tratamiento debido a reacciones adversas fue del 7% para ambos grupos de pacientes tratados con Benlysta y pacientes que recibieron placebo. Lista tabulada de reacciones adversas: Las reacciones adversas se enumeran a continuación según la clasificación de órganos del sistema MedDRA y por frecuencia. Las categorías de frecuencia utilizadas son: Muy frecuentes > 1/10; Frecuentes ≥1/100 a < 1/10; Poco frecuentes ≥1/1.000 a < 1/100. Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.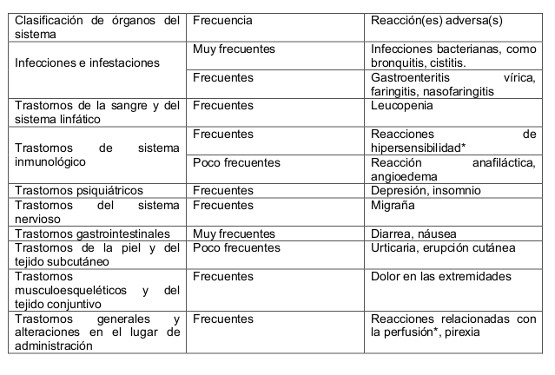
Descripción de reacciones adversas seleccionadas: Reacciones relacionadas con la perfusión y reacciones de hipersensibilidad: La incidencia de reacciones relacionadas con la perfusión y reacciones de hipersensibilidad que se produjeron durante o el mismo día de la perfusión fue 17% en el grupo que recibió Benlysta y 15% en el grupo que recibió placebo, de los que el 1% y 0,3%, respectivamente, requirieron interrupción permanente del tratamiento. Estas reacciones fueron observadas generalmente el día de la perfusión, pero las reacciones de hipersensibilidad aguda pueden producirse el día después de la administración. Los pacientes con antecedentes de alergias a múltiples medicamentos o con reacciones de hipersensibilidad significativa pueden tener mayor riesgo. Reacciones de hipersensibilidad clínicamente significativas con belimumab y que requieren discontinuación permanente fueron reportadas en 0.4% de los pacientes. Estas reacciones se observaron generalmente el día de la perfusión, y los pacientes con historia de múltiple alergia a drogas o reacciones de hipersensibilidad significativa pueden presentar un riesgo aumentado. Se ha observado retraso en el comienzo de las reacciones de hipersensibilidad agudas de varias horas luego de la perfusión y la recurrencia de reacciones clínicamente significativas después de una resolución inicial de los síntomas siguiendo el tratamiento apropiado también ha sido observada. Se han observado reacciones de hipersensibilidad no agudas del tipo retardado y han incluido síntomas tales como exantema, náusea, fatiga, mialgia, cefalea y edema. Infecciones: En estudios clínicos, la incidencia en el grupo que recibía belimumab fue del 70% comparado con el 67% en el grupo que recibía placebo. Las infecciones que se presentaron en al menos 3% de los pacientes que recibían belimumab y en al menos 1% más frecuentemente que los pacientes que recibían placebo fueron nasofaringitis, bronquitis, faringitis, cistitis y gastroenteritis viral. Infecciones serias se presentaron en 5% de los pacientes que recibieron tanto belimumab como placebo; infecciones oportunistas graves significaron < 1% y 0% de ellas, respectivamente. Algunas infecciones fueron graves o fatales. Se notificaron infecciones oportunistas en tres pacientes tratados con Benlysta. En un caso, la relación causal es improbable. No se notificaron infecciones oportunistas en el grupo placebo. Leucopenia: La incidencia de leucopenia notificada como un acontecimiento adverso fue del 4% en el grupo que recibió Benlysta y del 2% en el grupo que recibió placebo. Trastornos psiquiátricos: se produjo insomnio en el 7% del grupo que recibió Benlysta y 5% en el grupo placebo. Se notificaron casos de depresión en el 5% y 4% de los grupos que recibieron Benlysta y placebo, respectivamente. Trastornos gastrointestinales: Los pacientes obesos (IMC > 30 Kg/m2) tratados con Benlysta presentaron tasas más altas de náusea, vómitos y diarrea frente a placebo, y en comparación con los acontecimientos gastrointestinales en los pacientes obesos fue grave.
Advertencias.
Benlysta no se ha estudiado en los siguientes grupos de pacientes y no se recomienda su uso en: Lupus del sistema nervioso central activo grave; Nefritis lúpica activa grave; VIH; Pacientes con antecedentes o infección activa por virus de hepatitis B o C; Hipogammaglobulinemia (IgG < 400 mg/dl) o deficiencia de IgA (IgA < 10 mg/dl); Antecedentes de trasplante de órgano mayor o trasplante de células madre hematopoyéticas/médula ósea o trasplante renal. Uso concomitante con tratamientos dirigidos a las células B y ciclofosfamida: No se ha estudiado BENLYSTA en combinación con tratamientos dirigidos a las células B o con ciclofosfamida intravenosa. Se debe tener precaución si se coadministra BENLYSTA con otros tratamientos dirigidos a las células B o con ciclofosfamida. Hipersensibilidad y reacciones a la perfusión: La administración de BENLYSTA puede ocasionar reacciones de hipersensibilidad y a la perfusión. En caso de que se presente una reacción grave, debe interrumpirse la administración de BENLYSTA y administrarse un tratamiento médico apropiado. Pacientes con historia de alergias a múltiples drogas o significativa hipersensibilidad pueden tener un riesgo mayor (ver Reacciones adversas). Puede administrarse premedicación con un antihistamínico oral, con o sin un antipirético, antes de la perfusión de Belimumab. No hay evidencia suficiente para determinar si la premedicación disminuye la frecuencia o severidad de las reacciones a la perfusión. En estudios clínicos, las reacciones serias de hipersensibilidad a la perfusión afectaron a menos de 1% de los pacientes, e incluyeron reacción anafiláctica, bradicardia, hipotensión, angioedema y disnea. Las reacciones a la perfusión ocurrieron con mayor frecuencia en los dos primeros días de perfusión y tendieron a disminuir con las infusiones subsiguientes. Se ha notificado que los pacientes desarrollan síntomas de hipersensibilidad aguda varias horas después de la administración de la perfusión. Por tanto, Benlysta debe administrarse en un entorno que disponga de suficientes recursos para el manejo inmediato de estas reacciones. Los pacientes deben continuar bajo supervisión médica durante un período de tiempo prolongado (durante varias horas), al menos tras la administración de las dos primeras perfusiones, teniendo en cuenta la posibilidad de aparición de una reacción de inicio tardío. Se debe advertir a los pacientes que es posible que aparezcan reacciones de hipersensibilidad durante el día de la perfusión, o el día después, y se les debe informar de los signos y síntomas potenciales y de la posibilidad de recurrencia de los mismos. Se debe indicar a los pacientes que busquen atención médica inmediata si experimentan cualquiera de estos síntomas. Se debe proporcionar el prospecto al paciente siempre que se le administre Benlysta. Se han observado reacciones de hipersensibilidad no agudas del tipo retardado que incluyen síntomas como exantema, náusea, fatiga, mialgia, cefalea y edema facial. Riesgo de infecciones: Como con otros agentes inmunomoduladores, el mecanismo de acción de BENLYSTA puede aumentar el riesgo de desarrollar infecciones. Se han reportado infecciones graves, incluyendo casos fatales, en pacientes con SLE recibiendo terapia inmunosupresora, incluyendo BENLYSTA (ver Reacciones adversas). Los pacientes que desarrollen una infección al estar bajo tratamiento con BENLYSTA, deben ser monitoreados estrechamente, y debe considerarse la suspensión del inmunosupresor. Los médicos deben tener precaución al considerar el uso de BENLYSTA en pacientes con infecciones crónicas. Se desconoce el riesgo de utilizar Benlysta en pacientes con tuberculosis activa o latente. Leucoencefalopatía multifocal progresiva (PML por sus siglas en inglés): La leucoencefalopatía multifocal progresiva (PML) resultante en deficiencias neurológicas, incluyendo casos fatales, ha sido reportada en pacientes con LES que han recibido terapia inmunosupresora, incluyendo BENLYSTA. El diagnóstico de PML debe ser considerado en cualquier paciente con aparición de signos o síntomas de reciente comienzo o progresivos de deterioro neurológico. El paciente debe ser referido a un neurólogo o al especialista apropiado para su evaluación y si se confirma el diagnóstico de leucoencefalopatía multifocal progresiva, debe considerarse el suspender la terapia inmunosupresora, incluyendo BENLYSTA. Riesgo de malignidades: Como con otros agentes inmunomoduladores, el mecanismo de acción de BENLYSTA puede aumentar el riesgo potencial de desarrollar malignidades. En estudios clínicos, no se observó diferencia alguna en la tasa de malignidades entre los grupos tratados con BENLYSTA y los grupos tratados con placebo. Inmunización: No se deben administrar vacunas con microorganismos vivos 30 días antes, o de manera concurrente con BENLYSTA, ya que no se ha establecido su seguridad clínica. No hay datos disponibles acerca de la transmisión secundaria de infección de personas que reciben vacunas con microorganismos vivos a pacientes que están recibiendo BENLYSTA. Debido a su mecanismo de acción, BENLYSTA puede interferir con la respuesta a las inmunizaciones. Sin embargo, en un estudio para evaluar la respuesta a una vacuna 23 valente pneumocóccica, las respuestas globales inmunológicas a los diferentes serotipos fueron similares en pacientes con SLE recibiendo belimumab comparado con aquellos no recibiendo tratamiento en el momento de la vacunación. Neoplasias malignas y trastornos lipoproliferativos: Los medicamentos inmunomoduladores, incluyendo belimumab, pueden aumentar el riesgo de desarrollar neoplasias malignas. Se debe tener precaución cuando se considera el tratamiento de belimumab en pacientes con antecedentes de neoplasia maligna o cuando se continúa el tratamiento en pacientes que desarrollan neoplasia maligna. No se estudiaron pacientes con neoplasia maligna en los últimos 5 años, a excepción de aquellos con cáncer de células basales o de células escamosas de la piel, o cáncer de cuello de útero completamente extirpado o que habían sido tratados adecuadamente.
Interacciones.
No se han realizado estudios de interacción farmacológica con BENLYSTA. En estudios clínicos realizados en pacientes con LES, la administración concomitante de micofenolato mofetilo, azatioprina, hidroxicloroquina, metotrexato, antiinflamatorios no esteroidales, aspirina e inhibidores de la HMG CoA reductasa, no tuvo un efecto significativo en las exposiciones a Belimumab (ver Farmacocinética). Embarazo y lactancia: Fertilidad: No existen datos acerca de los efectos de BENLYSTA en la fertilidad humana. En estudios realizados en animales, no se han evaluado los efectos en la fertilidad masculina y femenina (ver Datos preclínicos de seguridad). Embarazo: Existen datos limitados acerca del uso de BENLYSTA en mujeres embarazadas. No se han realizado estudios formales. Los anticuerpos Inmunoglobulina G (IgG), incluyendo Belimumab, pueden atravesar la placenta. BENLYSTA debe utilizarse durante el embarazo sólo si el beneficio potencial justifica el riesgo potencial para el feto. Si se quiere garantizar la prevención del embarazo, las mujeres en edad reproductiva deben utilizar métodos anticonceptivos adecuados mientras se utiliza BENLYSTA y por lo menos cuatro meses después del último tratamiento con BENLYSTA. Los estudios realizados en animales no indicaron efectos perjudiciales directos o indirectos respecto a toxicidad materna, embarazo o desarrollo embriofetal. Los hallazgos relacionados con el tratamiento se limitaron a reducciones reversibles en el recuento de células B en monos lactantes (ver Datos preclínicos de seguridad). Se debe monitorear una disminución de células B en los lactantes de madres tratadas y dependiendo de los resultados, considerar el retrasar la vacunación con virus vivos. La reducción de células B en lactantes puede también interferir con la respuesta a las inmunizaciones (ver Advertencias). Lactancia: No se ha establecido la seguridad de BENLYSTA durante la lactancia. No existen datos acerca de la excreción de Belimumab en la leche humana, o de la absorción sistémica de Belimumab después de la ingestión. Aunque, Belimumab se excretó en la leche de monos macacos, la literatura publicada sugiere que el consumo de leche materna en neonatos humanos y lactantes no resulta en absorción clínicamente significativa de anticuerpos IgG maternos hacia la circulación. Se recomienda tomar una decisión acerca del tratamiento con BENLYSTA en madres en lactancia, tomando en cuenta la importancia de la lactancia para el lactante y la importancia del fármaco para la madre, y cualquier potential efecto adverso por belimumab para el lactante o por la condición materna subyacente. Efectos sobre la capacidad de conducir y utilizar máquinas: No se han realizado estudios para investigar el efecto de BENLYSTA en el desempeño para manejar o la capacidad de operar maquinaria. Con base en la farmacología de BENLYSTA, no se predicen efectos negativos en dichas actividades. Debe tenerse en mente el estado clínico del paciente y el perfil de seguridad de BENLYSTA al considerar la capacidad del paciente para realizar tareas que requieran juicio, habilidades motoras o cognitivas.
Incompatibilidades.
BENLYSTA no es compatible con dextrosa al 5%. BENLYSTA sólo debe prepararse y administrarse como se indica; véase Instrucciones de Uso / Manejo.
Conservación.
Viales cerrados: Almacenar entre 2°C y 8°C. No congelar. Proteger de la luz. Almacenar en la caja original hasta su uso. Solución reconstituida: Después de la reconstitución con agua inyectable, y de la dilución con solución el producto es estable hasta por 8 horas entre 2°C a 8°C. Protéjase de la luz solar. Vida de anaquel: La fecha de caducidad se indica en el empaque. Instrucciones de uso/manejo: Reconstitución y dilución: Belimumab no contiene conservadores; por lo tanto, la reconstitución y la dilución deben realizarse bajo condiciones asépticas. Permita que el vial se caliente a temperatura ambiente durante 10-15 minutos. Se recomienda usar una aguja calibre 21-25 para perforar el tapón del frasco para reconstituir y diluir. El vial monodosis de 120 mg de BENLYSTA debe reconstituirse con 1,5 mL de agua inyectable estéril, para lograr una concentración final de 80 mg/mL de Belimumab. El vial monodosis de 400 mg de BENLYSTA debe reconstituirse con 4,8 mL de agua inyectable estéril, para lograr una concentración final de 80 mg/mL de Belimumab. El flujo de agua estéril debe dirigirse hacia la pared del vial para minimizar la formación de espuma. Realice movimientos rotatorios suaves con el vial durante 60 segundos. Permita que el vial se asiente a temperatura ambiente durante la reconstitución, realizando movimientos rotatorios suaves con el vial durante 60 segundos cada 5 minutos hasta que el polvo se disuelva. No agite. La reconstitución normalmente está completa 10-15 minutos después de haber añadido el agua estéril, pero puede tomar hasta 30 minutos. Proteja la solución reconstituida de la luz. Si se utiliza un dispositivo mecánico de reconstitución para reconstituir BENLYSTA, no debe exceder 500 rpm y no se debe girar el vial por más de 30 minutos. Una vez que la reconstitución esté completa, la solución deberá ser opalescente e incolora a amarillo pálido, y sin partículas. Sin embargo, se esperan y son aceptables pequeñas burbujas de aire. El producto reconstituido se diluye a 250 mL con solución salina normal al 0,9% para perfusión IV. Las soluciones de dextrosa intravenosa al 5% son incompatibles con BENLYSTA y no deben utilizarse. De una bolsa o frasco de perfusión de 250 mL de solución salina normal, retire y elimine un volumen equivalente al volumen requerido de solución reconstituida de BENLYSTA para la dosis del paciente. Luego se agrega el volumen necesario de la solución reconstituida de BENLYSTA en la bolsa o frasco de perfusión. Invierta suavemente la bolsa o frasco para mezclar la solución. Se deberá desechar cualquier solución no utilizada en los viales. Inspeccione visualmente la solución de BENLYSTA en busca de partículas y decoloración antes de la administración. Deseche la solución si se observan partículas o decoloración. Si no se utiliza inmediatamente, se debe proteger la solución reconstituida de la luz solar directa y almacenarse en refrigeración entre 2°C a 8°C. Las soluciones diluidas en solución salina normal, pueden almacenarse entre 2°C a 8°C. El tiempo total desde la reconstitución de BENLYSTA hasta la finalización de la administración de la perfusión no debe exceder 8 horas. Administración: BENLYSTA debe administrarse en perfusión durante un período de 1 hora. BENLYSTA no debe administrarse concomitantemente en perfusión en la misma línea intravenosa con otros agentes. No se han realizado estudios físicos o bioquímicos de compatibilidad para evaluar la coadministración de BENLYSTA con otros agentes. No se han observado incompatibilidades entre BENLYSTA y las bolsas de cloruro de polivinilo o poliolefina.
Sobredosificación.
Hay experiencia clínica limitada con la sobredosis de BENLYSTA. Se han administrado dos dosis de hasta 20 mg/kg por perfusión intravenosa con 21 días de diferencia en humanos, sin observarse un incremento en la incidencia o severidad de las reacciones adversas, en comparación con dosis de 1, 4, o 10 m