BETAFERON
BAYER
Inmunomodulador.
Composición.
1 ml de solución reconstituida para inyección contiene 250 microgramos (8.0 millones de UI) de interferón recombinante beta-1b. Betaferon se ha formulado como un polvo estéril blanco o casi blanco y contiene 300 microgramos (9.6 millones de UI) de interferón recombinante beta-1b por vial, lo que incluye un excedente calculado del 20%. 1 ml de solución acuosa para reconstitución contiene 5,4 mg de cloruro sódico. Forma farmacéutica: Liofilizado para solución inyectable y solvente.
Farmacología.
Propiedades farmacodinámicas: Los interferones pertenecen a la familia de las citoquinas, que son proteínas naturales. Los interferones tienen pesos moleculares comprendidos entre 15.000 y 21.000 dalton. Se han identificado tres clases principales de interferones denominados alfa, beta y gamma. Los interferones alfa, beta y gamma tienen actividades biológicas diferentes, aunque se sobreponen en parte. Las actividades del interferón beta-1b están restringidas a la especie, y, por tanto, la información farmacológica de mayor interés es la que se deriva de los estudios realizados sobre cultivos de células humanas o los estudios in vivo en humanos. El interferón beta-1b ha demostrado poseer actividad antivírica y actividad inmunorreguladora. Los mecanismos mediante los cuales ejerce sus acciones en la esclerosis múltiple (EM) aún no están totalmente aclarados. Sin embargo, se sabe que las propiedades modificadoras de respuesta biológica del interferón beta-1b están mediadas por sus interacciones con receptores celulares específicos que se hallan en la superficie de las células humanas. El enlace del interferón beta-1b a estos receptores induce la expresión de un número de productos genéticos que se supone son los mediadores de las acciones biológicas del interferón beta-1b. Algunos de estos productos han sido determinados en el suero y en fracciones celulares de sangre obtenida de pacientes tratados con interferón beta-1b. El interferón beta-1b reduce la afinidad de enlace y aumenta la internalización y lisis del receptor de interferón. El interferón beta-1b también aumenta la actividad supresora de las células mononucleares de sangre periférica. No se han realizado ensayos específicos acerca de la influencia de Betaferon sobre el sistema cardiovascular, el sistema respiratorio ni sobre la función de órganos endocrinos. Los pacientes con la enfermedad secundaria progresiva tratados con Betaferon presentaron un retardo de hasta 12 meses en el tiempo de progresión de la discapacidad, incluyendo el tiempo hasta los estadios de discapacidad severa, es decir, pacientes que dependen de una silla de ruedas. Este retardo en la discapacidad se presentó en pacientes con o sin recaídas y en todos los niveles de discapacidad investigado (EDSS 3-6-5). Tanto los pacientes con esclerosis múltiple remitente recidivante como los con esclerosis múltiple secundaria progresiva tratados con Betaferon han mostrado una reducción en la frecuencia (30%) y severidad de las recaídas clínicas, así como una prolongación del intervalo libre de recaídas. Se redujo el número de hospitalizaciones y el uso de esteroides debidos a la enfermedad. Además, tanto en la esclerosis múltiple remitente recidivante como en la secundaria progresiva, Betaferon demostró un efecto beneficioso significativo sobre el número de lesiones medidas por medio de IRM en T2 ponderado y sobre las lesiones recientes activas medidas con IRM cada 6 semanas en la EM remitente recidivante y con IRM contrastada con Gd-DTPA en T1 ponderado cada mes (meses 1-6 y 19-24) en la EM secundaria progresiva. Se ha demostrado que el aumento del número de lesiones medidas por IRM se correlaciona con un aumento en la discapacidad medida con la escala ampliada del estado de discapacidad (EDSS). En el estudio de pacientes con un evento clínico único, los resultados informados por los pacientes, incluyendo la calidad de vida, fueron similares en los grupos Betaferon y placebo. Propiedades farmacocinéticas: Los niveles séricos del fármaco se determinaron en pacientes y voluntarios sanos mediante un bioensayo no completamente específico. Las concentraciones séricas de interferón beta-1b son bajas o no detectables después de la administración subcutánea de la dosis recomendada de 0.25 mg de Betaferon. Por lo tanto, no se dispone de la información farmacocinética en pacientes con EM en tratamiento con la dosis recomendada de Betaferon. Después de la inyección subcutánea de 0.5 mg de Betaferon a voluntarios sanso, se detectaron valores máximos en suero de aproximadamente 40 Ul/ml en el período de 1-8 horas después de la inyección. En este estudio, la biodisponibilidad absoluta de Betaferon en administración subcutánea fue aproximadamente del 50%. Los valores medios de depuración sérica y los valores de vida media de eliminación del suero, se han estimado a partir de varios estudios con administración intravenosa de interferón beta-1b, en no más de 30 ml/min/kg y de 5 horas respectivamente. Los inyecciones del producto en días alternos no dan lugar a elevación de la concentración sérica del fármaco y la farmacocinética tampoco parece modificarse durante el tratamiento. Después de la administración subcutánea de 0.25 mg de Betaferon en días alternos a voluntarios sanos, los niveles de marcadores de la respuesta biológica (neopterina, microglobulina-b2 y la citoquina inmunosupresiva IL-10) aumentaron significativamente por encima de los niveles basales en las 6 a 12 horas siguientes a la primera dosis de Betaferon. Los niveles de marcadores de la respuesta biológica alcanzaron su máximo entre las 40 y las 124 horas, y permanecieron por encima de los niveles basales durante todo el período experimental de siete días (168 horas). No se conoce la relación entre los niveles séricos de interferón beta-1b o los niveles de marcadores de la respuesta biológica inducid y el mecanismo por el que Betaferon ejerce su efecto sobre la EM. Datos preclínicos sobre seguridad: No se han realizado estudios de toxicidad aguda. Como los roedores no reaccionan al interferón beta humano, la elevación del riesgo se basó en los estudios de administración repetida efectuados en monos Rhesus. Se observó hipertermia transitoria, así como un aumento transitorio significativo de los linfocitos y un descenso transitorio significativo de los trombocitos y neutrófilos segmentados. No se han realizado estudios a largo plazo. Los estudios sobre reproducción en monos Rhesus revelaron toxicidad materna y una alta tasa de abortos. No se observaron malformaciones en los animales supervivientes. No se ha realizado ninguna investigación sobre la fertilidad. No se ha observado influencia alguna sobre el ciclo estral en monos. En un único estudio de genotoxicidad (test de Ames) no se observó efecto mutagénico. No se han realizado estudios de carcinogénesis. Un ensayo de transformación celular in vitro no dio indicio de potencial tumorigénico.
Indicaciones.
Betaferon está indicado para el tratamiento de: Esclerosis múltiple (EM) remitente recidivante y Esclerosis múltiple secundaria progresiva. En la esclerosis múltiple remitente recidivante Betaferon está indicado para la reducción de la frecuencia y la severidad de las recaídas clínicas en pacientes ambulatorios (es decir, pacientes capaces de caminar sin ayuda), caracterizadas por la aparición de, al menos, dos ataques de disfunción neurológica durante el período de los dos años anteriores, seguidos por recuperaciones completas o incompletas. En la esclerosis múltiple secundaria progresiva Betaferon está indicado para la reducción de la frecuencia y severidad de las recaídas clínicas y para el retardo de la progresión de la enfermedad.
Dosificación.
El tratamiento con Betaferon deberá iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de esta enfermedad. La dosis recomendada de Betaferon es de 250 microgramos (8 millones de UI), correspondiente a 1 ml de solución reconstituida (ver Instrucciones de uso/manipulación), inyectada por vía subcutánea cada 2 días. En general se recomienda al comienzo del tratamiento una titulación de la dosis. Los pacientes deben recibir inicialmente una dosis de 62.5 microgramos (0.25 ml) por vía subcutánea cada 2 días, la cual se incrementará lentamente hasta una dosis de 250 microgramos (1.0 ml) cada 2 días. Este período de titulación se podrá ajustar de acuerdo con la tolerancia individual.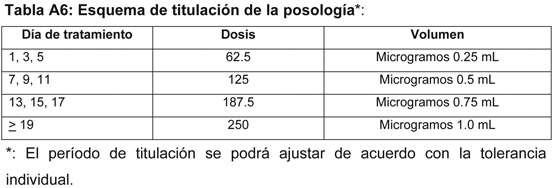
Actualmente no se conoce durante cuánto tiempo debe ser tratado el paciente. La eficacia durante un período de hasta 3 años ha quedado demostrada en un ensayo clínico controlado. Se dispone de datos de seguimiento bajo condiciones de ensayos clínicos controlados en pacientes con esclerosis múltiple (EM) remitente recidivante hasta por 5 años y para pacientes con EM secundaria progresiva hasta por 3 años. Existen datos no controlados de seguimiento para pacientes con EM secundaria progresiva hasta por 4.5 años. Para la EM remitente recidivante, los datos disponibles hasta por 5 años sugieren una eficacia sostenida del tratamiento con Betaferon durante todo el período de tiempo. En la EM secundaria progresiva se ha podido demostrar la eficacia del tratamiento bajo las condiciones controladas de los ensayos clínicos por un período de 2 años y, con datos limitados, por un período de hasta 3 años. No se ha investigado de manera sistemática la eficacia y seguridad de Betaferon en niños y adolescentes de menos de 18 años de edad. Por tanto, no deberá administrarse Betaferon en estas edades.
Contraindicaciones.
Betaferon está contraindicado en pacientes con historia de hipersensibilidad al interferón beta recombinante o natural, o a cualquiera de los excipientes. Betaferon está contraindicado en los siguientes casos: - Embarazo y lactancia. - Pacientes con historia de trastornos depresivos graves y/o ideación suicida. - Pacientes con hepatopatía descompensada.
Reacciones adversas.
Frecuentemente se ha observado sintomatología de tipo gripal (fiebre, escalofríos, artralgias, malestar, sudoración, cefalea o mialgias). La incidencia de estos síntomas decreció con el tiempo. En general, se recomienda al comienzo del tratamiento realizar una titulación de la dosis con el objeto de aumentar la tolerancia al Betaferon por parte del paciente (ver Dosificación). La sintomatología de tipo gripal puede reducirse también con la administración de medicamentos antiinflamatorios no esteroides. Tras la administración de Betaferon se observaron frecuentemente reacciones en el lugar de inyección. El tratamiento con 250 microgramos (8 millones de UI) de Betaferon se ha asociado de manera significativa a enrojecimiento, edema, decoloración, inflamación, dolor, hipersensibilidad, necrosis y a reacciones inespecíficas. La incidencia de reacciones en el lugar de la inyección disminuyó habitualmente con el tiempo. La incidencia de reacciones en el lugar de la inyección se puede reducir por medio del empleo de un autoinyector. La siguiente relación de eventos adversos y alteraciones de laboratorio está basada en los ensayos clínicos (tabla 1) y en la fármaco-vigilancia posterior a la comercialización de Betaferon (tabla 2). Es aún limitada la experiencia con Betaferon en pacientes con EM y, consecuentemente, pueden no haberse observado aún los efectos adversos de baja incidencia. Se anota el término MedDRA más apropiado para describir determinada reacción y sus sinónimos o condiciones relacionadas. En la tabla 1 se relacionan los eventos adversos y las alteraciones de laboratorio que se presentaron entre todos los pacientes tratados con 0.25 mg o 0.16 mg/m2 de Betaferon en días alternos por períodos de hasta 3 años en los ensayos clínicos controlados, con una incidencia que era por lo menos 2% mayor que aquella observada en los pacientes que recibieron placebo.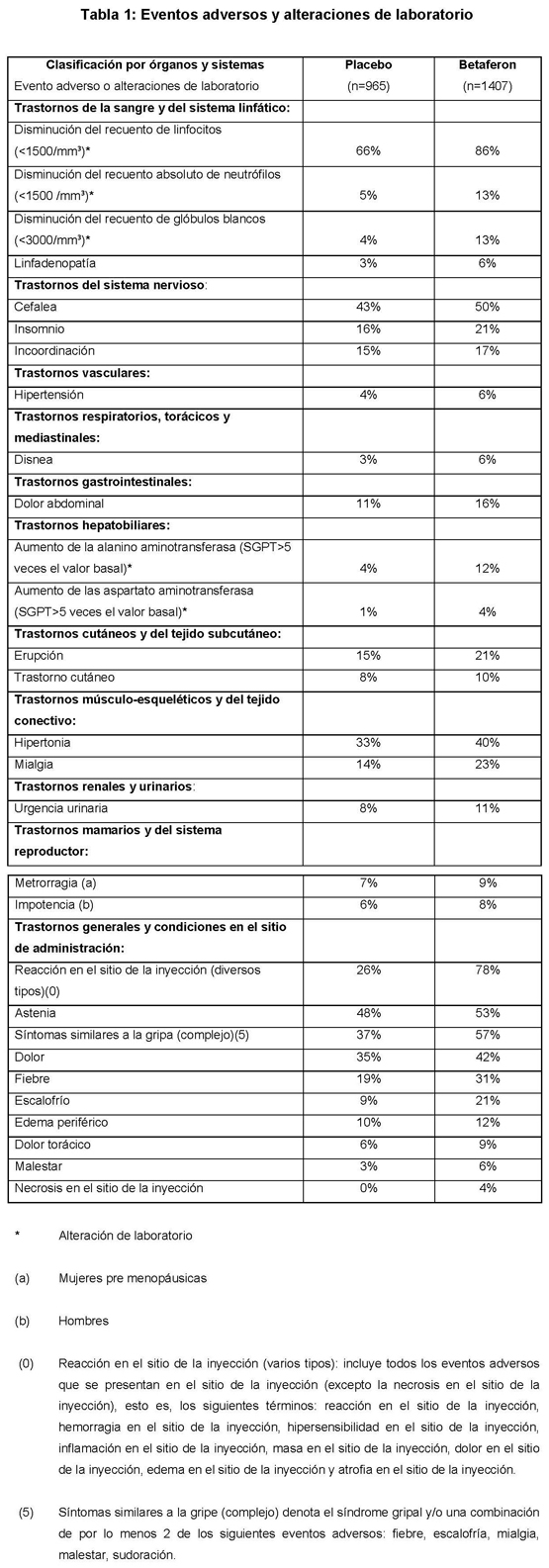
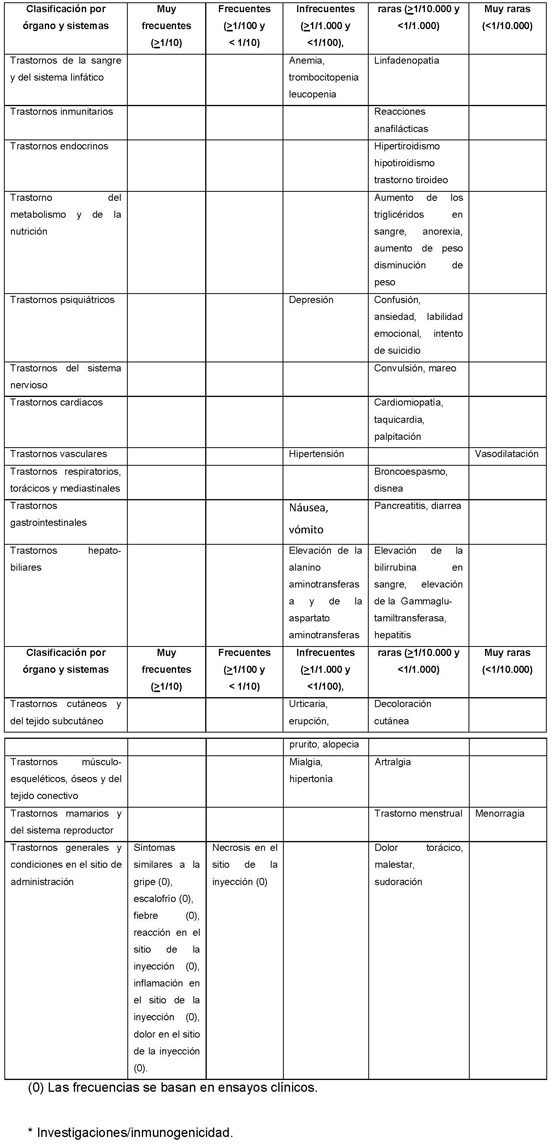
Como sucede con todas las proteínas de uso terapéutico, existe un potencial de inmunogenicidad. Durante los ensayos clínicos controlados se recolectaron muestras de suero cada 3 meses con el fin de vigilar el desarrollo de anticuerpos contra Betaferon. En los diferentes ensayos clínicos controlados entre 23% y un 41% de los pacientes desarrolló actividad sérica neutralizante contra el interferón beta-1b, confirmada por al menos dos títulos positivos consecutivos: de estos pacientes, entre un 43% y un 55% revirtió a un estado estable de anticuerpos negativos 8 con base en dos títulos consecutivos negativos) durante el período de observación subsecuente del estudio respectivo. No se ha demostrado un efecto atenuante constante sobre las medidas de impacto clínico, incluyendo hallazgos de IRM, en relación con la presencia de anticuerpos neutralizantes en los ensayos clínicos, puntos finales de análisis, diferentes análisis estadísticos y diversas definiciones del estado positivo de anticuerpos neutralizantes. No se ha establecido una asociación entre la presencia de eventos adversos y el desarrollo de una actividad neutralizante. La decisión de continuar o suspender el tratamiento debe basarse en consideraciones sobre la actividad clínica de la enfermedad y no en el estado de actividad neutralizante.
Advertencias.
Trastornos del sistema inmune: La administración de citoquinas a pacientes con una gammapatía monoclonal preexistente se ha asociado al desarrollo del síndrome de extravasación capilar sistémica con síntomas similares a los de un estado de shock y desenlace fatal. Trastornos gastrointestinales: En casos raros, se ha observado pancreatitis con el uso de Betaferon, a menudo asociada a hipertrigliceridemia. Trastornos del sistema nervioso: Antes del tratamiento con Betaferon los pacientes deben ser informados que pueden presentarse trastornos depresivos e ideación suicida como efectos secundarios del tratamiento y que estos síntomas deben comunicarse inmediatamente al médico que lo ha prescrito. Raramente estos síntomas pueden llevar un intento de suicidio. Los pacientes que presentan depresión e ideación suicida han de ser estrechamente vigilados y debe considerarse la interrupción del tratamiento. En dos ensayos clínicos controlados que incluían a 1657 pacientes con esclerosis múltiple secundaria progresiva no hubo diferencias significativas entre los pacientes tratados con Betaferon y los tratados con placebo con respecto a la depresión y a la ideación suicida. No obstante, dado que no se puede excluir que el tratamiento con Betaferon pueda estar asociado con la presencia de depresión y suicidio en casos individuales, Betaferon debe administrarse con precaución a pacientes que tengan o hayan tenido trastornos depresivos, o ideación suicida. Se debe considerar la suspensión del tratamiento con Betaferon si se presentan tales eventos durante la terapia. Este producto contiene albúmina humana y por lo tanto existe un riesgo extremadamente remoto para la transmisión de enfermedades virales. También se considera como extremadamente remoto el riesgo teórico de transmisión de la enfermedad de Creutzfeld-Jacob (CJD). Precauciones especiales: Pruebas de laboratorio: Además de aquellos exámenes de laboratorio que normalmente se requieren para el control de pacientes con esclerosis múltiple, antes de iniciar el tratamiento con Betaferon y a intervalos regulares después de iniciar el mismo se recomienda realizar un cuadro hemático completo, un recuento leucocitario diferencial, recuento plaquetario y química sanguínea, incluyendo pruebas de función hepática (por ej. AST (SGOT), ALT, (SGPT) así como (GT). Estas pruebas deben realizarse posteriormente de manera periódica en ausencia de síntomas clínicos. En pacientes con historia de enfermedades tiroideas o si está clínicamente indicado se recomienda la realización periódica de pruebas de función tiroidea. Pacientes con anemia, trombocitopenia y leucopenia (solas o en cualquier combinación) pueden requerir una vigilancia más estrecha de los recuentos completos de células sanguíneas, con recuentos diferenciales y plaquetarios. Trastornos hepatobiliares: Durante los ensayos clínicos con Betaferon se presentaron en los pacientes muy frecuentemente elevaciones asintomáticas de las aminotransferasas séricas, en su mayor parte de carácter leve y transitorio. Como ocurre con otros interferones beta, se ha reportado en raras ocasiones la presencia de daño hepático severo, incluyendo casos de insuficiencia hepática, en pacientes tratados con Betaferon. Los casos más severos se presentaron frecuentemente en pacientes expuestos a otros medicamentos o sustancias que se sabe están asociados con hepatotoxicidad, o en presencia de entidades médicas coexistentes (como por ej. enfermedades malignas con metástasis, infecciones severas, sepsis, y abuso de alcohol. Se debe vigilar la presencia de signos de lesión hepática. La aparición de elevaciones de las aminotransferasas séricas requiere una vigilancia estrecha y una investigación. Se debe considerar la suspensión de Betaferon si los niveles se elevan significativamente o si se asocian con signos clínicos tales como ictericia. En ausencia de evidencia clínica de daño hepático y después de la normalización de las enzimas hepáticas, se puede considerar la reintroducción del tratamiento con un seguimiento adecuado de las funciones hepáticas. Trastornos del sistema nervioso: Betaferon debe administrarse con precaución a pacientes que tengan antecedentes de convulsiones. Trastornos cardíacos: Betaferon se debe usar con precaución en pacientes con trastornos cardíacos preexistentes, por ej. En pacientes con insuficiencia cardíaca en estadio III/IV según la clasificación NYHA, porque estos pacientes fueron excluidos de los estudios clínicos, y en pacientes con miocardiopatía (ver Reacciones adversas). Se han reportado casos aislados de miocardiopatía: si esto ocurre y se sospecha una relación con Betaferon, se debe suspender el tratamiento. Trastornos generales y condiciones en el sitio de la inyección: Pueden presentarse reacciones graves de hipersensibilidad (reacciones agudas poco frecuentes pero severas, tales como broncoespasmo, anafilaxia y urticaria). Se han comunicado casos de necrosis en el lugar de la inyección en pacientes bajo tratamiento con Betaferon (ver Reacciones adversas). La necrosis puede ser extensa y abarcar la fascia muscular y el tejido graso con consecuente formación de cicatriz. Ocasionalmente, se requiere de desbridamiento y, con menor frecuencia aún, de injertos cutáneos. La curación puede demorar hasta 6 meses. Si el paciente experimenta alguna ruptura en la piel, la cual se puede asociar con edema o drenaje de líquido del lugar de inyección. Se le debe recomendar que consulte a su médico antes de continuar con las inyecciones de Betaferon. En caso de que el paciente tenga lesiones múltiples, se debe suspender el tratamiento con Betaferon hasta su curación. Los pacientes con lesiones únicas pueden continuar el tratamiento con Betaferon siempre que la necrosis no sea muy extensa, puesto que algunos pacientes han experimentado la curación de la necrosis en el lugar de la inyección sin tener que interrumpir el tratamiento. Para minimizar el riesgo de necrosis en el lugar de la inyección se debe aconsejar a los pacientes: Usar una técnica aséptica de inyección. Alternar los lugares de inyección con cada dosis. Se debe controlar periódicamente el procedimiento de autoinyección del paciente, especialmente si se han presentado reacciones en el lugar de la inyección.
Interacciones.
No se han realizado con Betaferon estudios específicos de interacción con medicamentos. No se conoce el efecto de la administración de 250 microgramos (8 millones de UI) de Betaferon, en días alternos, sobre el metabolismo de fármacos en pacientes de EM. El tratamiento de las recidivas con corticosteroides o ACTH durante períodos de hasta 28 días ha sido bien tolerado en pacientes que están recibiendo Betaferon. El empleo de Betaferon en administración conjunta con otros inmunomoduladores sólo se ha estudiado con corticoides o ACTH. Se ha comunicado que los interferones originan una reducción de la actividad de enzimas dependientes del citocromo hepática P450, tanto en animales como en seres humanos. Por ello, debe observarse precaución al administrar Betaferon en combinación con medicamentos que tengan un estrecho índice terapéutico y dependan notablemente para su depuración del sistema citocromo hepático P450. Se debe tener precaución con cualquier medicación concomitante que afecte al sistema hematopoyético. Embarazo y lactancia: Embarazo: Se desconoce si Betaferon es inocuo para el feto cuando se administra a la mujer gestante, o si puede afectar la capacidad reproductiva en humanos. Se han comunicado abortos espontáneos en pacientes con EM en ensayos clínicos controlados. El interferón beta-1b recombinante humano ha demostrado embriotoxicidad en monos Rhesus, causando una tasa elevada de abortos con dosis más elevadas (para los resultados preclínicos, véase Datos preclínicos sobre seguridad). Por ello, las mujeres fértiles deben tomar medidas anticonceptivas adecuadas. Si la paciente quedara embarazada, o tuviera intención de hacerlo durante el tratamiento con Betaferon, debe ser informada de los riesgos potenciales y se debe recomendar la interrupción del tratamiento (ver Resultados preclínicos). Lactancia: No se sabe si el interferón beta-1b se excreta en la leche materna. A causa de la posible inducción por Betaferon de reacciones adversas serias en los lactantes, debe decidirse si se interrumpe la lactancia o el tratamiento con el fármaco. Efectos sobre la capacidad de conducir vehículos o utilizar maquinaria: No ha sido investigado. Los efectos adversos sobre el SNC asociados al empleo de Betaferon podrían afectar la capacidad de conducir vehículos y utilizar maquinaria en pacientes susceptibles.
Incompatibilidades.
Ante la ausencia de estudios de compatibilidad, este producto medicina no debe mezclarse con otros medicamenta.
Sobredosificación.
Interferón beta-1b se ha administrado sin efectos adversos graves que comprometieran funciones vitales a pacientes adultos con cáncer, en dosis individuales de hasta 5.5 mg (176 millones de UI) por vía i.v., tres veces por semana.
Presentación.
Liofilizado para solución inyectable 8.000.000 U.I. con solvente.