BONDRONAT®
ROCHE
Bisfosfonato.
Composición.
Principio activo: sal monosódica del ácido ibandrónico monohidrato. Presentaciones de los viales:
Forma farmacéutica: Concentrado para solución para inyectable 6mg/ 6ml. Vía de administración: Intravenosa (i.v.). Declaración de esterilidad / radiactividad: No procede. Lista de excipientes: Cloruro sódico, ácido acético, acetato sódico, agua para inyectables.
Farmacología.
Propiedades farmacodinámicas: La acción farmacodinámica del ácido ibandrónico consiste en la inhibición de la resorción ósea. In vivo, el ácido ibandrónico previene la destrucción ósea inducida experimentalmente por el bloqueo de la función gonadal, retinoides, tumores o extractos tumorales. En ratas jóvenes (en crecimiento rápido) inhibe también la resorción ósea endógena, lo cual conduce a un aumento de la masa ósea en comparación con los animales no tratados. Los modelos animales confirman que el ácido ibandrónico es un inhibidor muy potente de la actividad osteoclástica. En ratas en crecimiento no se han observado indicios de trastornos de la mineralización ni siquiera con dosis 500 veces superiores a la necesaria para el tratamiento de la osteólisis de origen tumoral. En modelos animales de enfermedad metastásica ósea, el ácido ibandrónico no sólo previno el desarrollo de nuevas metástasis óseas, sino que también bloqueó la progresión de las ya formadas, inducidas por células de carcinoma de mama humano. También se demostraron efectos antitumorales directos en ratas atímicas ("desnudas") e in vitro. En el modelo de tumor intraóseo de Walker, el ácido ibandrónico inhibía totalmente la destrucción masiva de la integridad trabecular y del hueso cortical. El ácido ibandrónico también inhibía la hipercalciuria y la resorción ósea acelerada en el modelo subcutáneo. Estos efectos guardaban una relación directa con las variaciones de los marcadores urinarios de resorción ósea y de la cifra de osteoclastos. En todos los modelos, todos los efectos beneficiosos fueron más pronunciados cuando el tratamiento se inició lo antes posible, como se observó también en la reducción de las metástasis osteoscleróticas en el modelo de células MCF-7 de ratón. Las propiedades antimetastásicas cabe atribuirlas muy probablemente a una menor liberación mediada por osteoclastos de factores de crecimiento tumoral a partir de la matriz ósea. In vitro, el pretratamiento con ácido ibandrónico de cortes de hueso inhibía la fijación y difusión de células tumorales, así como la invasión de células tumorales. Cuando se agregó con células tumorales a cortes de hueso no tratados, el ácido ibandrónico tuvo efectos aditivos con compuestos citotóxicos como los taxoides. Los estudios clínicos en pacientes con enfermedad metastásica ósea han revelado un efecto inhibidor de la osteólisis en función de la dosis, expresado a través de marcadores de la resorción ósea, y un efecto en las complicaciones óseas dependiente también de la dosis. En los estudios clínicos en pacientes con hipercalcemia tumoral maligna se demostró que la acción inhibidora del ácido ibandrónico sobre la osteólisis de origen tumoral, y más concretamente sobre la hipercalcemia tumoral maligna, se caracteriza por un descenso del calcio sérico y de la excreción urinaria de calcio. Mecanismo de acción: El ácido ibandrónico es un bisfosfonato muy potente, perteneciente al grupo de los bisfosfonatos nitrogenados, que actúa sobre el tejido óseo e inhibe de forma específica la actividad osteoclástica, sin interferir en el reclutamiento de los osteoclastos. La acción selectiva del ácido ibandrónico sobre el tejido óseo obedece a su elevada afinidad por la hidroxiapatita, que constituye la matriz mineral ósea. El ácido ibandrónico reduce la resorción ósea, sin afectar directamente a la formación del tejido óseo. La resorción ósea de origen neoplásico se caracteriza por un exceso de resorción ósea no compensado por una adecuada formación de hueso. El ácido ibandrónico inhibe selectivamente la actividad osteoclástica reduciendo la resorción ósea y, de este modo, las complicaciones óseas de la enfermedad neoplásica. Ensayos clínicos / Eficacia: Tratamiento de la enfermedad metastásica ósea: En dos estudios clínicos de fase III aleatorizados y controlados con placebo, de 96 semanas, se evaluó Bondronat en comprimidos de 50 mg como tratamiento de la enfermedad metastásica ósea secundaria al carcinoma de mama. Pacientes de sexo femenino con carcinoma de mama y metástasis óseas radiológicamente confirmadas fueron asignadas aleatorizadamente a un grupo con placebo (277 pacientes) o a un grupo con Bondronat en comprimidos de 50 mg (287 pacientes). La tasa de períodos con morbididad esquelética (SMPR), una variable principal compuesta de valoración, se basó en la radioterapia ósea, la cirugía como tratamiento de fracturas y la incidencia de fracturas vertebrales y no vertebrales. Los datos agrupados de estos estudios demostraron una ventaja significativa de Bondronat sobre el placebo en la reducción de la SMPR. También disminuyó el riesgo de complicaciones óseas relacionadas (Skeletal Related Events, SRE) en las pacientes tratados con Bondronat frente a las que habían recibido placebo. A continuación se resumen los resultados de estos estudios. Asimismo, en un estudio de fase III aleatorizado y controlado con placebo, de 96 semanas y con iguales variables de valoración, se evaluó la administración i.v. de 6 mg de Bondronat. Pacientes de sexo femenino con carcinoma de mama y metástasis óseas radiológicamente confirmadas fueron asignadas aleatorizadamente a un grupo con placebo (158 pacientes) o a un grupo con Bondronat en comprimidos de 6 mg (154 pacientes). Los resultados se resumen en la tabla siguiente: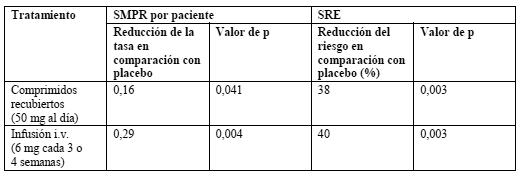
Las variables de valoración secundarias fueron el dolor óseo, la calidad de vida y los marcadores de resorción ósea en la orina. Bondronat puso de manifiesto una mejora de estos valores en comparación con placebo. En un estudio en 130 pacientes con carcinoma de mama metastásico se comparó la toxicidad de Bondronat administrado en infusión i.v. de 1 hora o de 15 minutos. No se apreciaron diferencias en los indicadores de la función renal. El perfil global de acontecimientos adversos del ácido ibandrónico tras la infusión de 15 minutos era compatible con el perfil de seguridad previamente descrito con infusiones de mayor duración, y no se identificaron nuevos problemas de toxicidad relacionados con la administración en infusión de 15 minutos. La infusión i.v. de 15 minutos no se ha estudiado en pacientes oncológicos con un aclaramiento de la creatinina inferior a 50 ml/min. Tratamiento de la hipercalcemia tumoral maligna: Bondronat en concentrado para solución para infusión: En los estudios clínicos se ha demostrado que la acción inhibidora del ácido ibandrónico sobre la osteólisis de origen tumoral, y más concretamente sobre la hipercalcemia tumoral maligna, se caracteriza por un descenso del calcio sérico y de la excreción urinaria del calcio. Con las dosis recomendadas para el tratamiento, en estudios clínicos con pacientes que presentaban una calcemia basal corregida con respecto a la albúmina igual o superior a 3,0 mmol/l se alcanzaron las tasas de respuestá indicadas a continuación, con los intervalos de confianza correspondientes, tras una rehidratación adecuada: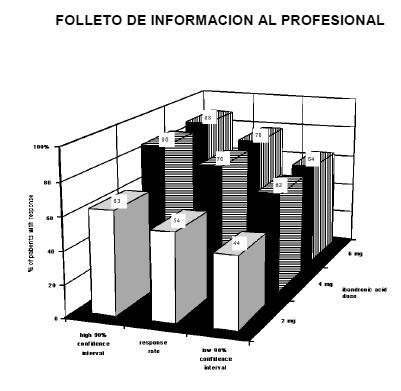
En estos pacientes y con esta dosis, la mediana de la duración hasta la normalización de la calcemia fue de 4 - 7 días. La mediana del tiempo hasta una recaída (nuevo aumento por encima de 3,0 mmol/l del calcio sérico corregido con respecto a la albúmina) fue de 18 a 26 días. Propiedades farmacocinéticas: Absorción: Administrado por vía oral, el ácido ibandrónico se absorbe rápidamente en los primeros tramos del tubo gastrointestinal. Las concentraciones plasmáticas máximas observadas se alcanzaron entre 0,5 y 2 h (mediana: 1 h) después de la dosis en ayunas. La biodisponibilidad absoluta era del orden del 0,6%. El grado de absorción disminuye cuando el ácido ibandrónico se administra con alimentos sólidos o líquidos (excepto el agua natural). Cuando el ácido ibandrónico se administra con un desayuno normal, su biodisponibilidad disminuye en torno al 90% con respecto a la alcanzada en ayunas. Si se administra 30 minutos antes de una comida, la reducción de la biodisponibilidad es del 30% aproximadamente. No se aprecia ninguna disminución importante de la biodisponibilidad cuando el ácido ibandrónico se administra 60 o más minutos antes de una comida. Las concentraciones plasmáticas de ácido ibandrónico son proporcionales a la dosis administrada hasta una dosis oral de 100 mg o una dosis intravenosa de 6 mg. La biodisponibilidad disminuía en torno al 75% cuando Bondronat en comprimidos se administraba 2 horas después de una comida normal. Por consiguiente, se recomienda tomar los comprimidos tras una noche en ayunas y continuar en ayunas durante otros 30 minutos, como mínimo, después de ingerida la dosis (v. Dosificación). Distribución: Tras la exposición sistémica inicial, el ácido ibandrónico se une rápidamente al hueso o se excreta con la orina. En el ser humano, el volumen terminal aparente de distribución es de 90 litros como mínimo, y la cantidad de fármaco que llega al tejido óseo se calcula en un 40 - 50% de la dosis circulante. La fijación a las proteínas plasmáticas es del orden del 87% con concentraciones terapéuticas, de modo que el riesgo de interacciones farmacológicas por desplazamiento es pequeño. Metabolismo: No hay indicios de que el ácido ibandrónico se metabolice en los animales o el ser humano. Eliminación: La fracción de ácido ibandrónico sistémicamente biodisponible desaparece de la circulación por absorción ósea (40 - 50%) y el resto se elimina de forma inalterada por los riñones. La fracción no absorbida de ácido ibandrónico se elimina inalterada con las heces. Se ha observado un amplio intervalo de valores de semivida aparente de eliminación en función de la dosis y la sensibilidad del análisis efectuado, pero la semivida terminal aparente suele situarse entre 10 y 60 horas. No obstante, la concentración plasmática inicial desciende hasta un 10% de los valores máximos en sólo 3 horas tras la administración intravenosa y 8 horas tras la administración oral. Después de 12 meses de administración oral diaria, la acumulación plasmática en pacientes con osteoporosis no se había duplicado. No se ha observado acumulación sistémica de ácido ibandrónico tras su administración intravenosa cada 4 semanas, durante 48 semanas, a pacientes con enfermedad metastásica ósea. El aclaramiento total del ácido ibandrónico es bajo, con valores medios que oscilan entre 84 y 160 ml/min. El aclaramiento renal (aproximadamente 60 ml/min en las mujeres posmenopáusicas sanas) representa el 50 - 60% del aclaramiento total y está relacionado con el aclaramiento de la creatinina. La diferencia entre el aclaramiento renal y el aclaramiento total aparente se atribuye a la captación ósea. Farmacocinética en poblaciones especiales: Sexo: La biodisponibilidad y la farmacocinética del ácido ibandrónico son similares en los hombres y las mujeres. Raza: No hay indicios de diferencias interétnicas de importancia clínica entre la población asiática y la caucasiana en cuanto a disposición del ácido ibandrónico. Los datos disponibles en pacientes de raza negra son muy escasos. Insuficiencia renal: La exposición al ácido ibandrónico en pacientes con diversos grados de insuficiencia renal guarda relación con el aclaramiento de la creatinina. En pacientes con insuficiencia renal grave (valor medio estimado de Acr: 21,2 ml/min) que habían recibido una dosis única de 2 mg (en infusión de 15 minutos), el valor medio de ABC0-24h se elevó en un 110% en comparación con el de voluntarios sanos. Tras la administración de una dosis única de 6 mg (en infusión de 15 minutos), el valor medio de ABC0-24h aumentó en un 14% en los pacientes con insuficiencia renal leve (valor medio estimado de Acr: 68,1 ml/min) y un 86% en los pacientes con insuficiencia renal moderada (valor medio estimado de Acr: 41,2 ml/min) en comparación con los sujetos con normofunción renal (valor medio estimado de Acr: 120 ml/min). La Cmáx media no aumentó en los pacientes con insuficiencia renal leve, y lo hizo en un 12% en los pacientes con insuficiencia renal moderada. No es necesario ajustar la dosis en los pacientes con insuficiencia renal leve (Acr entre 50 y 80 ml/min). En caso de insuficiencia renal moderada (Acr entre 30 y 50 ml/min) o grave (Acr < 30 ml/min) en pacientes con carcinoma de mama y metástasis óseas tratadas para prevenir complicaciones óseas, se recomienda ajustar la dosis (v. Farmacocinética en poblaciones especiales): En una sesión estándar de hemodiálisis de 4 horas se elimina del organismo alrededor del 37% del ácido ibandrónico. Insuficiencia hepática: No existen datos farmacocinéticos disponibles sobre el ácido ibandrónico en pacientes con insuficiencia hepática. El hígado no desempeña ninguna función importante en el aclaramiento del ácido ibandrónico, puesto que éste no se metaboliza sino que desaparece de la sangre por excreción renal y por captación ósea. Por lo tanto, no es necesario ajustar la dosis en los pacientes con insuficiencia hepática. Además, dado que la fijación del ácido ibandrónico a las proteínas plasmáticas es baja en concentraciones terapéuticas (85%), es poco probable que la hipoproteinemia de las hepatopatías graves pueda dar lugar a aumentos clínicamente importantes de la concentración plasmática de fármaco libre. Ancianos: En un análisis estadístico multifactorial, la edad no fue un factor independiente para ninguna de las variables farmacocinéticas estudiadas. Dado que la función renal disminuye con la edad, tal es el único factor que debe tomarse en consideración. Niños: No existen datos sobre el uso de Bondronat en niños y adolescentes menores de 18 años. Datos preclínicos sobre seguridad: Como con otros bisfosfonatos, se ha identificado el riñón como el órgano preferente de toxicidad sistémica. En los animales se han observado cambios renales clínicamente importantes cuando la exposición era más de tres veces superior a la máxima humana (datos basados en la concentración plasmática máxima tras administración intravenosa), lo cual indica que el margen de seguridad en el uso clínico es suficientemente amplio. Carcinogenicidad: No se ha observado ningún indicio de potencial carcinógeno. Mutagenicidad: No se ha observado ningún indicio de potencial genotóxico. Trastornos de la fertilidad: En los estudios de la fertilidad, el ácido ibandrónico alteraba la fertilidad de las ratas hembra en dosis de 1,2 mg/kg/día i.v. y reducía el número de lugares de implantación en dosis de 1,0 - 16,0 mg/kg/día p.o. y 1,2 mg/kg/día i.v. Teratogenicidad: En ratas y conejos no se han observado indicios de toxicidad fetal directa o efectos teratógenos del ácido ibandrónico administrado por vía intravenosa u oral. Otros efectos: Los efectos adversos del ácido ibandrónico en los estudios de toxicidad reproductiva en ratas han sido los previstos dentro del grupo de los bisfosfonatos. Tales son la disminución del número de sitios de implantación, la interferencia con el parto natural (distocia), el aumento de las variaciones viscerales (síndrome ureteropielorrenal) y anomalías en la dentición de la generación F1 en las ratas.
Indicaciones.
Bondronat está indicado para: Prevención de acontecimientos óseos (fracturas patológicas, complicaciones óseas que requieran radioterapia o cirugía) en pacientes con cáncer de mamas o metástasis ósea. Tratamiento de la hipercalcemia secundaria a un tumor.
Dosificación.
Dosis habitual: Tratamiento de la enfermedad metastásica ósea: Administración intravenosa: La dosis recomendada para la enfermedad metastásica ósea es de 6 mg i.v. cada 3 - 4 semanas. La dosis debe administrarse en infusión de 15 minutos como mínimo. Para preparar la infusión, se debe agregar el contenido de los viales a una solución salina isotónica o una solución glucosada al 5%. Ver Pautas posológicas especiales para instrucciones detalladas sobre la administración intravenosa en la enfermedad metastásica ósea. Tratamiento de la hipercalcemia tumoral maligna: Antes del tratamiento con Bondronat debe rehidratarse adecuadamente al paciente con solución salina al 0,9%. Ha de tenerse en cuenta la gravedad de la hipercalcemia y el tipo de tumor. Por regla general, los pacientes con metástasis osteolíticas necesitan dosis menores que los pacientes con hipercalcemia de origen humoral. En la mayoría de los pacientes con hipercalcemia grave (calcio sérico corregido por la albúmina* ≥ 3 mmol/l o ≥ 12 mg/dl) basta con una dosis única de 4 mg. En los pacientes con hipercalcemia moderada (calcio sérico corregido por la albúmina < 3 mmol/l o < 12 mg/dl), 2 mg es una dosis eficaz. La dosis máxima administrada en los estudios clínicos ha sido de 6 mg, pero esta dosis no es más eficaz. *Nota: Calcio sérico corregido por la albúmina (mmol/l) = calcio sérico (mmol/l) - [0,02 x albúmina (g/l)] + 0,8 o Calcio sérico corregido por la albúmina (mg/dl) = calcio sérico (mg/dl) + 0,8 x [4 - albúmina (g/dl)]. Para convertir el valor de calcemia corregida con respecto a la albúmina de mmol/l en mg/dl, multiplíquese por 4. En la mayor parte de los casos, la concentración sérica de calcio puede normalizarse en un plazo de 7 días. La mediana del tiempo hasta la recaída (nuevo aumento por encima de 3 mmol/l del calcio sérico corregido por la albúmina) ha sido de 18 - 19 días con las dosis de 2 y 4 mg. La mediana del tiempo hasta la recaída ha sido de 26 días con la dosis de 6 mg. A un número limitado de pacientes (n=50) se les ha administrado una segunda infusión contra la hipercalcemia. En caso de hipercalcemia recurrente o de eficacia insuficiente del tratamiento, puede considerarse la posibilidad de repetir éste. Bondronat en concentrado para solución para infusión debe administrarse en infusión i.v. Para ello, el contenido de los viales se agrega a 500 ml de solución salina isotónica (o a 500 ml de solución glucosada al 5%) y se administra a lo largo de 1 - 2 horas. Pautas posológicas especiales: Insuficiencia hepática: No es probable que sea necesario ajustar la dosis (ver Farmacocinética en poblaciones especiales). Insuficiencia renal: Concentrado para solución para infusión en pacientes con enfermedad metastásica ósea: No es necesario ajustar la dosis en los pacientes con insuficiencia renal leve (Acr entre 50 y 80 ml/min). En caso de insuficiencia renal moderada (Acr entre 30 y 50 ml/min) o grave (Acr < 30 ml/min) en pacientes con carcinoma de mama y metástasis óseas tratadas para prevenir complicaciones óseas, se recomienda seguir las recomendaciones posológicas siguientes (ver Farmacocinética en poblaciones especiales):
La infusión i.v. de 15 minutos no se ha estudiado en pacientes oncológicos con un aclaramiento de la creatinina inferior a 50 ml/min. Ancianos: No es necesario ajustar la dosis. Niños: No se han determinado la seguridad y la eficacia en pacientes menores de 18 años.
Contraindicaciones.
Bondronat en viales con concentrado para solución para infusión está contraindicado en pacientes con: hipocalcemia (ver Advertencias); alergia al ácido ibandrónico o a cualquiera de los excipientes.
Reacciones adversas.
Ensayos clínicos: Tratamiento de la hipercalcemia tumoral maligna: La Tabla 1 recoge los efectos adversos observados en los estudios clínicos (n=352 pacientes) en los que se utilizó Bondronat por vía intravenosa para tratar la hipercalcemia tumoral maligna. Se agruparon los datos de los estudios clínicos en los que se habían administrado dosis de 2 mg y 4 mg de ácido ibandrónico. Los efectos adversos se registraron sin determinación de la causalidad.
Tratamiento de la enfermedad metastásica ósea: En la Tabla 2 se muestran las reacciones adversas observadas con una frecuencia ≥ 5% en el estudio fundamental de fase III, notificadas como remota, posible o probablemente relacionadas con Bondronat en una dosis de 6,0 mg i.v. a intervalos de 4 semanas. Se han excluido de la tabla los acontecimientos adversos notificados con igual frecuencia en ambos grupos de pacientes (tratamiento activo y placebo) y los notificados con mayor frecuencia en el grupo de placebo.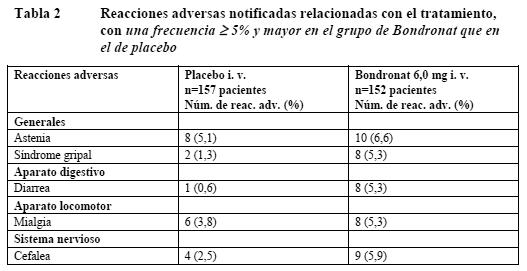
Experiencia tras la comercialización: Hasta la fecha, no se han notificado nuevos datos de la farmacovigilancia tras la comercialización con la administración intravenosa de Bondronat que aporten nueva información al perfil de seguridad de este medicamento. Trastornos musculoesqueléticos y del tejido conjuntivo: En pacientes tratados con ácido ibandrónico se ha descrito osteonecrosis de la mandíbula en muy raras ocasiones (ver Advertencias). Trastornos oculares: Con bisfosfonatos, el ácido ibandrónico incluido, se han descrito episodios de inflamación ocular, como uveítis, episcleritis y escleritis. En algunos casos, la resolución de estos episodios no se produjo hasta la retirada del bisfosfonato. Trastornos del sistema inmunitario: Se han descrito casos de reacción anafiláctica/shock, incluidos algunos de desenlace fatal, en pacientes tratados con ácido ibandrónico (ver Advertencias).
Advertencias.
Advertencias y precauciones generales: Bondronat no debe administrarse a niños, ya que no existe experiencia clínica en este grupo de edad. Antes de iniciar el tratamiento con Bondronat, es preciso corregir la hipocalcemia y otros trastornos del metabolismo óseo y mineral. En todos los pacientes es importante una ingesta suficiente de calcio y vitamina D. Los pacientes deben recibir suplementos de calcio o vitamina D cuando sea insuficiente la ingesta de estos nutrientes con la alimentación. Puede producirse hipocalcemia, en cuyo caso ha de corregirse debidamente la concentración sérica de calcio. Considerando que tanto la administración accidental por vía intraarterial de preparaciones no recomendadas expresamente para este fin como la administración paravenosa pueden provocar lesiones hísticas, se tomarán las debidas precauciones para garantizar la administración intravenosa de Bondronat en concentrado para solución para infusión. Se han descrito casos de reacción anafiláctica/shock, incluidos algunos de desenlace fatal, en pacientes tratados con ácido ibandrónico i.v. Cuando se administre Bondronat por vía i.v., deben estar preparadas medidas adecuadas de ayuda y vigilancia médica. En caso de reacciones anafilácticas u otro tipo de reacciones de hipersensibilidad/alérgicas, se ha de retirar inmediatamente la infusión e instaurar el tratamiento adecuado. Se ha descrito osteonecrosis de la mandíbula (ONM) en pacientes tratados con bisfosfonatos. La mayoría eran pacientes con cáncer sometidos a procedimientos dentales, pero en algunos casos se trataba de pacientes con osteoporosis posmenopáusica u otros diagnósticos. Entre los factores de riesgo conocidos se hallan el diagnóstico de cáncer, tratamientos concomitantes (por ejemplo: quimioterapia, radioterapia, corticosteroides) y trastornos de comorbididad (por ejemplo: anemia, coagulopatía, infección, enfermedad dental preexistente). Los casos notificados han sido en su mayor parte de pacientes tratados con bisfosfonatos por vía intravenosa, pero en algunos casos fue oral la vía de administración. En los pacientes que experimenten ONM durante el tratamiento con bisfosfonatos, la cirugía dental puede empeorar su estado. Para los pacientes que requieran un procedimiento dental, no existen datos que indiquen si la suspensión del tratamiento con bisfosfonatos reduce el riesgo de ONM. El juicio clínico del médico debe guiarle en su actuación terapéutica con cada paciente de acuerdo con la valoración individual del balance de riesgos y beneficios. Estudios clínicos aleatorizados y controlados con placebo en pacientes con enfermedad metastásica ósea secundaria a carcinoma de mama no han revelado ningún indicio de deterioro de la función renal con el uso de Bondronat a largo plazo. No obstante, en función del estado clínico individual del paciente, se recomienda vigilar la función renal, así como el calcio, el fosfato y el magnesio séricos en los pacientes tratados con Bondronat. Si existe riesgo de insuficiencia cardíaca, ha de evitarse la hiperhidratación. Ver Pautas posológicas especiales: Concentrado para solución para infusión en pacientes con enfermedad metastásica ósea. Efectos sobre la capacidad para conducir y utilizar máquinas: No se han llevado a cabo estudios sobre los efectos en la capacidad de conducir vehículos y utilizar maquinaria.
Interacciones.
Interacciones farmacológicas: No se han observado interacciones con la administración simultánea de Bondronat y melfalano/prednisolona a pacientes con mieloma múltiple. Otros estudios de interacciones en mujeres posmenopáusicas han demostrado la ausencia de potencial de interacción con el tamoxifeno o la estrogenoterapia sustitutiva. En los estudios clínicos, Bondronat se ha administrado junto con antineoplásicos, diuréticos, antibióticos y analgésicos de uso habitual sin que se observaran interacciones clínicas. En voluntarios sanos de sexo masculino y mujeres posmenopáusicas, la ranitidina i.v. aumentó la biodisponibilidad del ácido ibandrónico en torno al 20% (es decir, dentro del intervalo normal de biodisponibilidad del ácido ibandrónico), probablemente como resultado de la disminución de la acidez gástrica. Sin embargo, no es necesario ajustar la dosis de Bondronat cuando se administre con antihistamínicos H2 u otros fármacos que eleven el pH gástrico. Por lo que respecta a la disposición, no son probables interacciones farmacológicas de importancia clínica. El ácido ibandrónico se elimina exclusivamente por excreción renal y no sufre ningún tipo de biotransformación. Parece que la vía de excreción no incluye los conocidos sistemas de transporte ácido o básico implicados en la excreción de otros fármacos. Además, el ácido ibandrónico no inhibe las principales enzimas hepáticas humanas del sistema P450, ni induce este sistema enzimático en las ratas. La fijación a las proteínas plasmáticas es baja en concentraciones terapéuticas, de modo que es poco probable que el ácido ibandrónico desplace a otras sustancias con actividad farmacológica. Uso en poblaciones especiales: Embarazo: Embarazo: categoría C: Bondronat no debe utilizarse durante el embarazo. En los estudios de la fertilidad, el ácido ibandrónico alteraba la fertilidad de las ratas hembra en dosis de 1,2 mg/kg/día i.v. y reducía el número de lugares de implantación en dosis de 1,0 - 16,0 mg/kg/día p.o. y 1,2 mg/kg/día i.v. En dosis de 5,0 - 20 mg/kg/día p.o. y 0,05 - 0,5 mg/kg/día i.v., el ácido ibandrónico interfería en el parto natural (distocias). En las ratas tratadas con ácido ibandrónico no hubo indicios de efectos tóxicos -teratógenos inclusive- en los fetos. Se registró un aumento de las variaciones viscerales (síndrome pieloureteral) en las ratas tratadas con dosis de 10 - 100 mg/kg/día p.o. o 1 mg/kg/día i.v. de ácido ibandrónico. En los conejos tratados con ácido ibandrónico no se observaron efectos teratógenos, embriotóxicos ni fetotóxicos con dosis de hasta 20 mg/kg/día p.o. o 0,07 mg/kg/día i.v. No existe experiencia clínica sobre el uso de Bondronat en mujeres embarazadas. Lactancia: Se desconoce si Bondronat pasa a la leche materna humana. Los estudios en ratas lactantes han demostrado la presencia de concentraciones bajas de ácido ibandrónico en la leche tras la administración intravenosa. Bondronat no debe utilizarse durante la lactancia. Uso en geriatría: Ver Pautas posológicas especiales y Farmacocinética en poblaciones especiales. Insuficiencia renal: Ver Pautas posológicas especiales y Farmacocinética en poblaciones especiales. Insuficiencia hepática: Ver Pautas posológicas especiales y Farmacocinética en poblaciones especiales.
Conservación.
Concentrado para solución para infusión: La solución para infusión con el producto reconstituido permanece física y químicamente estable durante 24 horas (no debe conservarse a más de 25°C). Desde un punto de vista microbiológico, la solución para infusión intravenosa debe utilizarse inmediatamente. Si no se utiliza inmediatamente, la duración y las condiciones de conservación del producto antes de su utilización son de la responsabilidad del usuario. Habitualmente, no debería sobrepasar las 24 horas a 2 - 8°C, salvo que la reconstitución se haya realizado en condiciones asépticas controladas y validadas. Instrucciones especiales de uso, manipulación y eliminación: El concentrado para solución para infusión es para uso en monodosis exclusivamente. Utilícese únicamente si la solución está límpida y no contiene partículas. Se recomienda respetar estrictamente la vía intravenosa de administración del concentrado para solución para infusión de Bondronat. Para prevenir posibles incompatibilidades, el concentrado para solución para infusión de Bondronat sólo debe diluirse con solución isotónica de cloruro sódico o con solución glucosada al 5%. Soluciones que contengan calcio no deben mezclarse con el concentrado para solución para infusión de Bondronat. Los restos de solución no utilizada deben desecharse. Eliminación de medicamentos no utilizados/caducados: La emisión de productos farmacéuticos al medio ambiente debe reducirse a un mínimo. Los medicamentos no deben eliminarse a través de las aguas residuales, y su eliminación con los residuos domésticos también debe evitarse. Utilice los sistemas de recogida establecidos si los hay en su localidad.
Sobredosificación.
Hasta el presente no se ha notificado ningún caso de sobredosis de Bondronat (administrado por vía oral o intravenosa). No existe información específica sobre el tratamiento de sobredosis de Bondronat. Ahora bien, sobredosis orales podrían causar efectos secundarios de tipo digestivo alto, como malestar gástrico, pirosis, esofagitis, gastritis o úlcera gástrica. Se pueden tomar leche o antiácidos para fijar Bondronat. Dado el riesgo de irritación esofágica, se desaconseja la provocación del vómito y se aconseja mantener al paciente en posición totalmente erecta. Las técnicas de hemodiálisis convencionales comportan un aclaramiento significativo del ácido ibandrónico.
Presentación.
Viales de 6 ml: envases con 1 y 5.