BONVIVA®
ROCHE
Bonviva es un bisfosfonato nitrogenado.
Composición.
Principio activo: Ácido ibandrónico, sal monosódica, monohidrato. Solución inyectable: Cada jeringa precargada contiene 3,375 mg de sal monosódica del ácido ibandrónico monohidrato, equivalentes a 3 mg de ácido ibandrónico, en 3 ml de solución. Forma farmacéutica: Jeringas precargadas. Vía de administración: Intravenosa (i.v.). Declaración de esterilidad / radiactividad: Producto estéril. Lista de excipientes: Cloruro de sodio Ph. Eur./USP. Ácido acético glacial Ph. Eur./USP. Acetato de sodio Ph. Eur./USP. Agua para inyectables. Ph. Eur./USP.
Farmacología.
Propiedades farmacodinámicas: La acción farmacodinámica del ácido ibandrónico consiste en la inhibición de la resorción ósea. In vivo, el ácido ibandrónico previene la destrucción ósea inducida experimentalmente por bloqueo de la función gonadal, retinoides, tumores o extractos tumorales. En ratas jóvenes (en crecimiento rápido) inhibe también la resorción ósea endógena, lo cual conduce a un aumento de la masa ósea en comparación con animales no tratados. Los modelos animales confirman que el ácido ibandrónico es un inhibidor muy potente de la actividad osteoclástica. En ratas en fase de crecimiento no se han observado indicios de alteración de la mineralización ni siquiera con dosis 5.000 veces superiores a la dosis necesaria para el tratamiento de la osteoporosis. La administración prolongada diaria o intermitente (con largos intervalos sin medicación) a ratas, perros y monos se asoció con la neoformación de tejido óseo de calidad normal y/o un aumento de la resistencia mecánica incluso con dosis superiores a las previstas para el uso terapéutico, también dentro del intervalo de toxicidad. En la especie humana, la eficacia de la administración oral de ácido ibandrónico diaria o intermitente con un intervalo sin medicación de 9 - 10 semanas se confirmó en un estudio clínico (MF 4411) en el que Bonviva demostró su eficacia contra las fracturas. Tanto las dosis diarias o intermitentes (con un intervalo sin medicación de 9 - 10 semanas por trimestre) como la administración i.v. de Bonviva a mujeres posmenopáusicas indujeron cambios bioquímicos indicativos de una inhibición de la resorción ósea dependiente de la dosis. La inyección i.v. de Bonviva reducía la concentración sérica de CTX dentro de los 3 - 7 días siguientes al comienzo del tratamiento, así como las cifras de osteocalcina dentro de los 3 meses siguientes. Tras suspender el tratamiento, se apreciaba un regreso a los valores patológicos preterapéuticos de elevación de la resorción ósea característicos de la osteoporosis posmenopáusica. Los análisis histológicos de biopsias óseas después de dos y tres años de tratamiento de mujeres posmenopáusicas con Bonviva en dosis de 2,5 mg diarios y dosis i.v. intermitentes de hasta 1 mg cada 3 meses revelaban un hueso de calidad normal, sin ningún signo de mineralización defectuosa. Mecanismo de acción: El ácido ibandrónico es un bisfosfonato muy potente, perteneciente al grupo de los bisfosfonatos nitrogenados, que actúa sobre el tejido óseo e inhibe de forma específica la actividad osteoclástica, sin interferir en el reclutamiento de los osteoclastos. La acción selectiva del ácido ibandrónico sobre el tejido óseo obedece a su elevada afinidad por la hidroxiapatita, que constituye la matriz mineral ósea. El ácido ibandrónico reduce la resorción ósea, sin afectar directamente a la formación del tejido óseo. En las mujeres posmenopáusicas disminuye el recambio óseo acelerado hacia niveles premenopáusicos, con lo que se produce un aumento neto progresivo de la masa ósea. La administración diaria o intermitente de ácido ibandrónico disminuye la resorción ósea, como reflejan la reducción de las concentraciones sérica y urinaria de los marcadores bioquímicos de recambio óseo, el aumento de la densidad mineral ósea (DMO) y el descenso de la incidencia de fracturas. Ensayos clínicos / Eficacia: Tratamiento de la osteoporosis posmenopáusica: Bonviva en una dosis diaria de 2,5 mg: En un estudio clínico (MF 4411) de doble-ciego, aleatorizado y controlado con placebo, de tres años de duración, se demostró una disminución estadísticamente significativa y médicamente importante en la incidencia de nuevas fracturas vertebrales radiográficas, morfométricas y clínicas. Bonviva se estudió en dosis orales de 2,5 mg diarios y 20 mg de forma intermitente (20 mg en días alternos hasta completar 12 dosis al comienzo de cada ciclo de 3 meses, separados por sendos períodos de reposo farmacológico de 9 - 10 semanas). Bonviva se administró 60 minutos antes del primer alimento (líquido o sólido) del día (período de ayuno postadministración). En el estudio participaron 2.946 mujeres de 55 a 80 años (en 2.928 de ellas era evaluable la eficacia), con 5 años como mínimo de posmenopausia, una DMO de 2 - 5 DE por debajo de la media premenopáusica (puntuación T) en al menos una vértebra lumbar (L1 - L4) y antecedentes de una a cuatro fracturas vertebrales. Todas las pacientes recibieron diariamente 500 mg de calcio y 400 UI de vitamina D. En ambos grupos de tratamiento con Bonviva se demostró una reducción estadísticamente significativa y médicamente importante de la incidencia de nuevas fracturas vertebrales. Con la pauta de 2,5 mg diarios, la incidencia de nuevas fracturas vertebrales radiográficas disminuyó en un 62% a lo largo de los tres años del estudio, mientras que las fracturas vertebrales clínicas disminuyeron en un 49%. Este potente efecto en las fracturas vertebrales se reflejó asimismo en una disminución estadísticamente significativa de la pérdida de estatura en comparación con placebo. Este efecto protector contra las fracturas fue constante durante todo el estudio, y no se apreciaron indicios de desvanecimiento del efecto con el paso del tiempo. Aunque este estudio clínico no estaba específicamente diseñado para demostrar la eficacia del ácido ibandrónico en las fracturas extravertebrales, en un subgrupo de pacientes con alto riesgo de fractura pudo comprobarse una reducción del riesgo relativo de fracturas extravertebrales (DMO en el cuello femoral: puntuación T < - 3,0 DE) de magnitud similar (69%) a la observada para las fracturas vertebrales. La observación de eficacia en las fracturas extravertebrales de subgrupos de alto riesgo concuerda bien con los resultados de los estudios clínicos realizados con otros bisfosfonatos. Con la pauta de administración diaria, la DMO lumbar aumentó en un 5,3% con respecto al placebo al cabo de los tres años del estudio; en comparación con los valores iniciales, el aumento fue del 6,5%. Los marcadores bioquímicos de recambio óseo (como CTX urinario y osteocalcina sérica) pusieron de manifiesto el patrón previsto de inhibición hasta niveles premenopáusicos, con valores máximos de inhibición al cabo de 3 - 6 meses. Tan sólo un mes después de iniciado el tratamiento con Bonviva se observó ya una reducción del 50% (con la pauta de 2,5 mg diarios) y del 78% (con la pauta de 20 mg de forma intermitente) en los marcadores bioquímicos de resorción ósea, en ambos casos de importancia clínica. La disminución de los marcadores bioquímicos de resorción ósea fue ya evidente en la primera semana de tratamiento. Bonviva en inyección de 3 mg cada 3 meses: Densidad mineral ósea (DMO): Bonviva en inyección i.v. de 3 mg cada 3 meses demostró ser al menos tan eficaz como en una dosis oral diaria de 2,5 mg en un estudio multicéntrico, aleatorizado, de doble-ciego y de no inferioridad de 2 años (BM 16549) en mujeres posmenopáusicas (1.386; edad: 55 - 80 años) con osteroporosis (T de DMO basal en la columna lumbar: < -2,5 DE). Todas las pacientes recibieron diariamente un suplemento de 400 UI de vitamina D y 500 mg de calcio. Este efecto se demostró tanto en el análisis principal después de un año como en el análisis confirmatorio después de dos años (Tabla 3). Los resultados del análisis principal de los datos del estudio BM 16550 al cabo de un año y del análisis confirmatorio demostraron la no inferioridad del régimen de una inyección de 3 mg cada 3 meses frente al de una dosis oral diaria de 2,5 mg en cuanto a incremento medio de la DMO en la columna lumbar, la cadera, el cuello femoral y el trocánter (Tabla 3).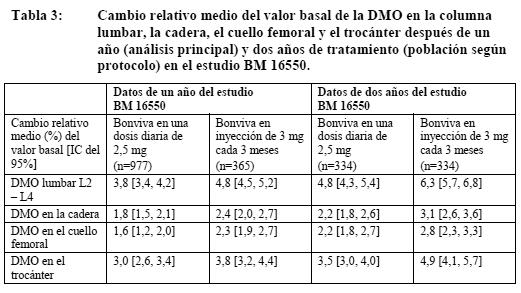
En un análisis prospectivo programado después de un año (p=0,001) y dos años (p=0,001), Bonviva en una inyección de 3 mg cada 3 meses fue superior a Bonviva en una dosis oral diaria de 2,5 mg para aumentar la DMO lumbar. Por lo que respecta a la DMO lumbar, en el 92,1% de las pacientes tratadas con una inyección de 3 mg cada 3 meses había aumentado o se mantenía la DMO después de 1 año (es decir, eran respondedoras), frente al 84,9% de las que recibían 2,5 mg diarios por vía oral (p=0,002). Después de 2 años de tratamiento, en el 92,8% de las pacientes tratadas con inyecciones de 3 mg y el 84,7% de las tratadas con dosis orales de 2,5 mg se mantenía o había aumentado la DMO lumbar (p=0,01). En cuanto a la DMO en la cadera, el 82,3% de las pacientes tratadas con una inyección de 3 mg cada 3 meses eran respondedoras al cabo de un año, frente al 75,1% de las que recibían 2,5 mg diarios por vía oral (p=0,02). Después de 2 años de tratamiento, en el 85,6% de las pacientes tratadas con inyecciones de 3 mg y el 77,0% de las tratadas con dosis orales de 2,5 mg se mantenía o había aumentado la DMO en la cadera (p=0,004). La proporción de pacientes cuya DMO tanto en la columna lumbar como en la cadera había aumentado o se mantenía al cabo de 1 año era del 76,2% en el grupo de 3 mg i.v. cada 3 meses y del 67,2% en el grupo de 2,5 mg por vía oral diarios (p=0,007). Al cabo de dos años cumplían este criterio el 80,1% y el 68,8% de las pacientes tratadas con 3 mg cada 3 meses y 2,5 mg al día, respectivamente (p=0,001). Marcadores bioquímicos del recambio óseo: En todas las determinaciones realizadas se hallaron reducciones clínicamente significativas del CTX sérico. Después de 12 meses, la mediana de los cambios relativos de los valores basales fueron del -58,6% con el régimen de una inyección i.v. de 3 mg cada 3 meses y del -62,6% con el régimen de 2,5 mg diarios por vía oral. Además, el 64,8% de las pacientes tratadas con 3 mg i.v. cada 3 meses cumplían los criterios de respondedora (descenso > 50% del valor basal), frente al 64,9% de las tratadas con 2,5 mg al día por vía oral. La reducción del CTX sérico se mantuvo durante 2 años, cumpliendo más de la mitad de las pacientes de ambos grupos los criterios de respondedora. Considerando los resultados del estudio BM 16550, se espera que Bonviva en una inyección i.v. de 3 mg cada 3 meses sea al menos tan eficaz como en una dosis oral diaria de 2,5 mg para prevenir las fracturas. Propiedades farmacocinéticas: Los efectos farmacológicos del ácido ibandrónico no guardan una relación directa con la concentración plasmática real, puesto que el lugar de acción se halla en el hueso. Así se ha observado en estudios realizados tanto con animales como en el ser humano, en los cuales se demostró una eficacia equivalente del ácido ibandrónico en administración diaria o intermitente, con un intervalo de descanso farmacológico de varias semanas (como mínimo 6 semanas en las ratas, 11 semanas en los perros, 30 días en los monos, 9,5 semanas en el ser humano), siempre y cuando se administrara la misma dosis total durante este período. La concentración plasmática de ácido ibandrónico es proporcional a la dosis administrada por vía intravenosa entre 0,5 mg y 6 mg. Distribución: Tras la exposición sistémica inicial, el ácido ibandrónico se une rápidamente al hueso o se excreta con la orina. En el ser humano, el volumen terminal aparente de distribución es de 90 litros como mínimo, y la cantidad de fármaco que llega al tejido óseo se calcula en un 40 - 50% de la dosis circulante. La fijación a las proteínas plasmáticas es baja (aproximadamente del 85% en concentraciones terapéuticas), de modo que el riesgo de interacciones farmacológicas por desplazamiento es pequeño. Metabolismo: No hay indicios de que el ácido ibandrónico se metabolice en los animales o el ser humano. Eliminación: Tras la administración intravenosa, el ácido ibandrónico desaparece de la circulación por absorción ósea (40 - 50%) y el resto se elimina de forma inalterada por los riñones. El intervalo de valores de semivida aparente de eliminación depende de la dosis y la sensibilidad analítica, pero la semivida terminal aparente suele situarse entre 10 y 72 horas. La concentración plasmática inicial desciende rápidamente hasta un 10% de los valores máximos en el espacio de 3 horas tras la administración intravenosa u oral. El aclaramiento total del ácido ibandrónico es bajo, con valores medios que oscilan entre 84 y 160 ml/min. El aclaramiento renal (aproximadamente 60 ml/min en las mujeres posmenopáusicas sanas) representa el 50 - 60% del aclaramiento total y está relacionado con el aclaramiento de la creatinina. La diferencia entre el aclaramiento renal y el aclaramiento total aparente se atribuye a la captación ósea. Farmacocinética en poblaciones especiales: Sexo: La farmacocinética del ácido ibandrónico es similar en los hombres y las mujeres. Raza: No hay indicios de diferencias interétnicas con importancia clínica entre los asiáticos y las personas de raza blanca en cuanto a disposición del ácido ibandrónico. Los datos disponibles sobre pacientes de raza negra son escasos. Insuficiencia renal: El aclaramiento renal del ácido ibandrónico guarda una relación lineal con el aclaramiento de la creatinina (Acr) en pacientes con diversos grados de insuficiencia renal. No es necesario ajustar la dosis en las pacientes con insuficiencia renal o hepática leve o moderada (Acr ≥ 30 ml/min). Tras la administración i.v. de 0,5 mg, el aclaramiento disminuía en un 67% (aclaramiento total), un 77% (aclaramiento renal) y un 50% (aclaramiento extrarrenal) en los pacientes con insuficiencia renal grave. El consiguiente aumento de la exposición no comportaba, sin embargo, una reducción de la tolerabilidad. Insuficiencia hepática: No existen datos farmacocinéticos disponibles sobre el ácido ibandrónico en pacientes con insuficiencia hepática. El hígado no desempeña ninguna función importante en el aclaramiento del ácido ibandrónico, que no se metaboliza, sino que desaparece de la sangre mediante excreción renal y captación ósea. Por lo tanto, no es necesario ajustar la dosis en los pacientes con insuficiencia hepática. Además, dado que la fijación del ácido ibandrónico a las proteínas plasmáticas es baja (85%) en concentraciones terapéuticas, parece poco probable que la hipoproteinemia de las hepatopatías graves pueda dar lugar a aumentos clínicamente importantes de la concentración plasmática de fármaco libre. Ancianos: Dado que el ácido ibandrónico no se metaboliza, cabe suponer que la única diferencia en la eliminación entre pacientes ancianos y jóvenes obedezca a cambios en la función renal relacionados con la edad (ver Insuficiencia renal en Pautas posológicas especiales). No es necesario ajustar la dosis por razones de edad. Niños: No existen datos sobre el uso de Bonviva en niños y adolescentes menores de 18 años. Datos preclínicos sobre seguridad: En los animales de experimentación, efectos tóxicos sólo se observaron con exposiciones consideradas suficientemente superiores a la exposición humana máxima, lo que era indicativo de poca trascendencia para el uso clínico. Carcinogenicidad: No se han observado indicios de riesgo carcinógeno o genotóxico.
Indicaciones.
Bonviva está indicado como tratamiento de la osteoporosis posmenopáusica para reducir el riesgo de fracturas. Tratamiento de la osteoporosis: La osteoporosis puede confirmarse por una masa ósea reducida (T < -2,0 DE [desviación estándar]) y la presencia o antecedentes de fracturas osteoporóticas, o bien por una masa ósea reducida (T < -2,5 DE) en ausencia de fracturas osteoporóticas preexistentes documentadas.
Dosificación.
La dosis recomendada de Bonviva es de 3 mg en inyección i.v (administrada en 15-30 segundos), cada tres meses. Las pacientes han de recibir suplementos de calcio y vitamina D. En caso de saltarse una dosis, debe procederse a la inyección tan pronto como sea razonablemente posible. Las inyecciones siguientes se efectuarán cada 3 meses, contados desde la fecha de la última inyección. Pautas posológicas especiales: Insuficiencia hepática: No es necesario ajustar la dosis (ver Farmacocinética en poblaciones especiales). Insuficiencia renal: No es necesario ajustar la dosis en las pacientes con valores de creatinina sérica ≤ 200 mmol/l (2,3 mg/dl) o de aclaramiento de la creatinina (determinado o estimado) ≥ 30 ml/min. No se recomienda Bonviva en inyección i.v. de 3 mg cada 3 meses para pacientes con cifras de creatinina sérica > 200 mmol/l (2,3 mg/dl) o con un aclaramiento de la creatinina (determinado o estimado) < 30 mg/ml, puesto que no hay datos clínicos de estudios en tales pacientes (ver Farmacocinética en poblaciones especiales). Ancianos: No es necesario ajustar la dosis. Niños: No se han determinado la seguridad y la eficacia en pacientes menores de 18 años.
Contraindicaciones.
Bonviva está contraindicado en pacientes con antecedentes de alergia al ácido ibandrónico o a alguno de los excipientes. Bonviva en inyección i.v. de 3 mg cada 3 meses está contraindicado en pacientes con hipocalcemia no corregida.
Reacciones adversas.
Ensayos clínicos: Tratamiento de la osteoporosis posmenopáusica: Administración oral de 2,5 mg al día: La seguridad de Bonviva en una dosis diaria de 2,5 mg se evaluó en 1.251 pacientes tratadas en 4 estudios clínicos controlados con placebo; el 73% de ellas procedía del estudio fundamental (pivotal) de tratamiento de tres años (MF 4411). En todos estos estudios, el perfil global de la seguridad de Bonviva en una dosis diaria de 2,5 mg fue similar al registrado con placebo. La proporción global de pacientes con reacciones adversas, es decir, acontecimientos adversos posible o probablemente relacionados con la medicación en estudio, fue, respectivamente, del 19,8% y el 17,9% con Bonviva y placebo en el estudio fundamental de tratamiento (MF 4411). Administración i.v. de 3 mg cada 3 meses: En el estudio fundamental de dos años en mujeres posmenopáusicas con osteoporosis (BM 16550), la seguridad global de Bonviva en inyección i.v. de 3 mg cada 3 meses y en una dosis oral diaria de 2,5 mg fue similar. La proporción global de pacientes con reacciones medicamentosas adversas al cabo de uno y dos años fue, respectivamente, del 26,0% y el 28,6% con Bonviva en inyección de 3 mg cada tres meses y del 20,4% y el 22,6% con Bonviva en una dosis oral diaria. La mayoría de las reacciones adversas fueron de intensidad leve o moderada, y en la mayor parte de los casos no obligaron a suspender el tratamiento. Las tablas 1 y 2 recogen los acontecimientos adversos después de uno y dos años, respectivamente, de tratamiento que se registraron en el estudio fundamental de fase III BM 16550 como posible o probablemente relacionados con la medicación en estudio en más del 1% de las pacientes tratadas con Bonviva en inyección i.v. de 3 mg cada 3 meses o placebo en inyección i.v. + Bonviva en una dosis oral diaria de 2,5 mg. No se incluyen las reacciones adversas notificadas en las pacientes tratadas con Bonviva en inyección de 3 mg cuya frecuencia era igual o inferior a la registrada en las tratadas por vía oral. Las Tablas 1 y 2 recogen las reacciones medicamentosas adversas en las pacientes tratadas durante 3 años con 2,5 mg diarios de Bonviva oral en el estudio de la eficacia contra las fracturas (MF 4411). De ambos estudios se muestran las reacciones medicamentosas adversas con una incidencia superior en las pacientes tratadas con Bonviva que en las que recibieron placebo en el estudio MF 4411. Dentro de cada grupo de frecuencia, los efectos adversos se presentan en orden decreciente de gravedad.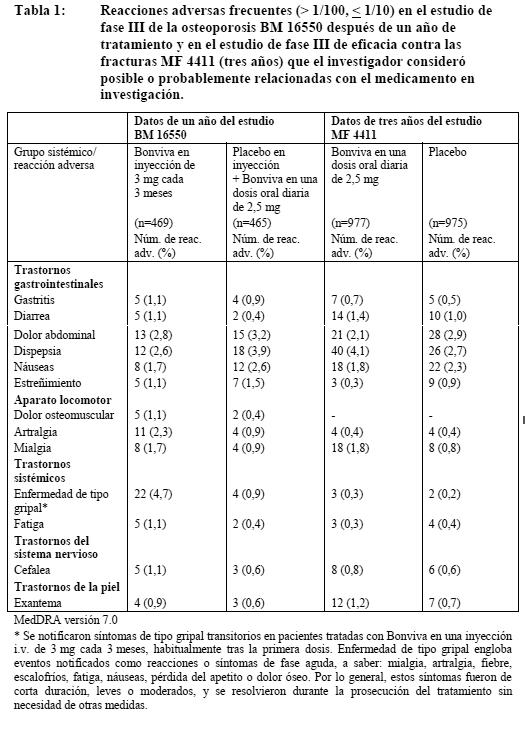
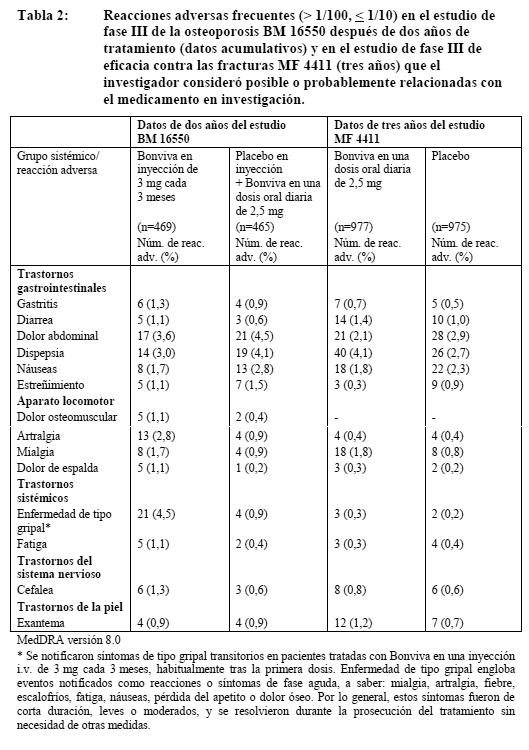
Reacciones medicamentosas adversas con una frecuencia ≤ 1% en el estudio BM 16650 que el investigador consideró posible o probablemente relacionadas con el medicamento en investigación. Dentro de cada grupo de frecuencia, los efectos adversos se presentan en orden decreciente de gravedad. Nada frecuentes ( > 1/1.000 y < 1/100): Aparato locomotor: Dolor óseo. Trastornos generales y reacciones en el sitio de administración: Astenia. Reacciones en la zona de inyección: Trastornos vasculares: Flebitis/tromboflebitis. Escasos ( > 1/10.000 y 1/1.000): Trastornos del sistema inmunitario: Reacciones de hipersensibilidad. Trastornos de la piel y del tejido subcutáneo: Edema angioneurótico. Hinchazón de cara/edema. Urticaria. Alteraciones analíticas: No se observaron indicios de que Bonviva en inyección i.v. de 3 mg cada 3 meses indujera alteraciones analíticas indicativas de disfunción hepática o renal, alteración del sistema hemático, hipocalcemia o hipofosfatemia. Experiencia tras la comercialización: Trastornos musculoesqueléticos y del tejido conjuntivo: En pacientes tratados con ácido ibandrónico se ha descrito osteonecrosis de la mandíbula en muy raras ocasiones (ver Advertencias). Trastornos oculares: Con bisfosfonatos, el ácido ibandrónico incluido, se han descrito episodios de inflamación ocular, como uveitis, episcleritis y escleritis. En algunos casos, la resolución de estos episodios no se produjo hasta la retirada del bisfosfonato. Trastornos del sistema inmunitario: Se han descrito casos de reacción anafiláctica/shock, incluidos algunos de desenlace fatal, en pacientes tratados con ácido ibandrónico (ver Advertencias).
Advertencias.
Advertencias y precauciones generales: Como otros bisfosfonatos administrados por vía i.v., Bonviva puede reducir transitoriamente los valores de calcio sérico. Antes de iniciar el tratamiento i.v. con Bonviva, es preciso corregir la hipocalcemia u otros trastornos del metabolismo óseo y mineral. En todas las pacientes es importante que el consumo de calcio y vitamina D sea suficiente. Las pacientes han de recibir suplementos de calcio y vitamina D. No se recomienda Bonviva en inyección i.v. de 3 mg cada 3 meses para pacientes con cifras de creatinina sérica > m200 mol/l (2,3 mg/dl) o con un aclaramiento de la creatinina (determinado o estimado) < 30 mg/ml, puesto que no hay datos clínicos de estudios en tales pacientes (ver Farmacocinética en poblaciones especiales). En caso de enfermedad o medicación simultánea que pueda tener efectos renales adversos, se mantendrá un control regular de la paciente durante el tratamiento de acuerdo con las buenas prácticas médicas. Se han descrito casos de reacción anafiláctica/shock, incluidos algunos de desenlace fatal, en pacientes tratados con ácido ibandrónico i.v. Cuando se administre Bonviva en inyección i.v., deben estar preparadas medidas adecuadas de ayuda y vigilancia médica. En caso de reacciones anafilácticas u otro tipo de reacciones de hipersensibilidad/alérgicas, se ha de suspender inmediatamente la inyección e instaurar el tratamiento adecuado. Se prestará cuidado a no administrar Bonviva por vía intraarterial o paravenosa, ya que se podrían dañar los tejidos. Se ha descrito osteonecrosis de la mandíbula (ONM) en pacientes tratados con bisfosfonatos. La mayoría eran pacientes con cáncer sometidos a procedimientos dentales, pero en algunos casos se trataba de pacientes con osteoporosis posmenopáusica u otros diagnósticos. Entre los factores de riesgo conocidos se hallan el diagnóstico de cáncer, tratamientos concomitantes (por ejemplo: quimioterapia, radioterapia, corticosteroides) y trastornos de comorbilidad (por ejemplo: anemia, coagulopatía, infección, enfermedad dental preexistente). Los casos notificados han sido en su mayor parte de pacientes tratados con bisfosfonatos por vía i.v., pero en algunos la vía de administración fue la oral. En los pacientes que experimenten ONM durante el tratamiento con bisfosfonatos, la cirugía dental puede empeorar su estado. Para los pacientes que requieran un procedimiento dental, no existen datos que indiquen si la suspensión del tratamiento con bisfosfonatos reduce el riesgo de ONM. El juicio clínico del médico debe guiarle en su actuación terapéutica con cada paciente de acuerdo con la valoración individual del balance de riesgos y beneficios. Efectos sobre la capacidad para conducir y utilizar máquinas: No se han llevado a cabo estudios sobre los efectos en la capacidad de conducir vehículos y utilizar maquinaria.
Interacciones.
Interacciones farmacológicas: En relación con la disposición, no es probable ninguna interacción de importancia clínica, puesto que el ácido ibandrónico no inhibe las principales enzimas hepáticas humanas del sistema P450 y tampoco induce este sistema enzimático en las ratas. Además, la fijación a las proteínas plasmáticas es baja en concentraciones terapéuticas, por lo que no parece probable que el ácido ibandrónico desplace a otros fármacos. El ácido ibandrónico se elimina exclusivamente por excreción renal y no sufre ningún tipo de biotransformación. Parece que la vía de excreción no incluye los conocidos sistemas de transporte ácido o básico implicados en la excreción de otros fármacos. En los estudios de interacciones farmacocinéticas en mujeres posmenopáusicas se ha demostrado la ausencia de potencial de interacción con el tamoxifeno y la estrogenoterapia restitutiva. No se ha observado ninguna interacción cuando el ácido ibandrónico se ha coadministrado con melfalán/prednisolona a pacientes con mieloma múltiple. Uso en poblaciones especiales: Embarazo: Bonviva no debe administrarse durante el embarazo y la lactancia. Embarazo: No se han realizado estudios específicos con el régimen de administración trimestral. En los estudios con una pauta de administración i.v. diaria del ácido ibandrónico, no se observó ni en las ratas ni en los conejos ningún indicio de toxicidad directa para el feto o de efectos teratógenos. El incremento ponderal disminuyó en la generación F1 de las ratas. Otros efectos adversos del ácido ibandrónico en los estudios de toxicidad reproductiva en ratas fueron idénticos a los observados con los bisfosfonatos como grupo farmacológico. Entre ellos se cuentan la disminución del número de sitios de implantación, la interferencia con el parto natural (distocia) y el aumento de variaciones viscerales (síndrome ureteropielorrenal). No existe experiencia clínica sobre el uso de Bonviva en mujeres embarazadas. Lactancia: Lactancia: En las ratas lactantes tratadas con 0,08 mg/kg/día de ácido ibandrónico por vía i.v., la concentración máxima de ácido ibandrónico en la leche materna fue de 8,1 ng/ml y se registró en las 2 primeras horas tras la administración i.v. Al cabo de 24 h, las concentraciones en la leche y en el plasma eran similares y correspondían a un 5%, aproximadamente, de la concentración medida a las 2 h de administrada la dosis. Se ignora si Bonviva pasa a la leche materna humana. Uso en pediatría: Ver Farmacocinética en poblaciones especiales, Niños. Uso en geriatría: Ver Farmacocinética en poblaciones especiales, Ancianos. Insuficiencia renal: Ver Farmacocinética en poblaciones especiales, Insuficiencia renal. Insuficiencia hepática: Ver Farmacocinética en poblaciones especiales, Insuficiencia hepática.
Incompatibilidades.
Bonviva no debe mezclarse con soluciones que contengan calcio ni con otros fármacos de administración intravenosa.
Estabilidad.
Este medicamento sólo deberá utilizarse hasta la fecha de caducidad, indicada con "VEN" en el envase.
Conservación.
No debe conservarse a más de 30°C. Instrucciones especiales de uso, manipulación y eliminación: Las jeringas son de un solo uso (monodosis). Utilícense únicamente jeringas en las que la solución esté límpida y sin partículas. Se recomienda respetar estrictamente la vía intravenosa de administración. Si el producto se administra mediante la técnica "piggyback" a través de un tubo de infusión i.v., ésta debe limitarse a una solución salina isotónica o glucosada al 5%. La solución inyectable no utilizada debe eliminarse, junto con la jeringa y la aguja, de acuerdo con la normativa local pertinente. Eliminación de jeringas/objetos cortopunzantes: En cuanto a la utilización y eliminación de jeringas y otros objetos médicos cortopunzantes, han de observarse estrictamente las siguientes medidas: No reutilizar nunca agujas ni jeringas. Depositar todas las agujas y jeringas utilizadas en un recipiente para objetos cortopunzantes (recipiente imperforable desechable). Mantener este recipiente fuera del alcance de los niños. En la medida de lo posible, no eliminar los recipientes de objetos cortopunzantes usados con los desechos domésticos. Eliminar los recipientes llenos conforme a la normativa local o las indicaciones del proveedor de servicios sanitarios. En caso de uso doméstico, debe entregarse a los pacientes un recipiente imperforable para eliminar las jeringas y agujas usadas. Eliminación de medicamentos no utilizados/caducados: La emisión de productos farmacéuticos al medio ambiente debe reducirse a un mínimo. Los medicamentos no deben eliminarse a través de las aguas residuales, y su eliminación con los residuos domésticos también debe evitarse. Utilice los sistemas de recogida establecidos si los hay en su localidad.
Sobredosificación.
No hay información específica sobre el tratamiento de una sobredosis de Bonviva. Considerando los conocimientos sobre este grupo farmacológico, una sobredosis i.v. puede originar hipocalcemia, hipofosfatemia e hipomagnesemia. Descensos clínicamente significativos de los valores séricos de calcio, fósforo o magnesio deben corregirse administrando por vía i.v. gluconato de calcio, fosfato de potasio o sodio y sulfato de magnesio, respectivamente.
Presentación.
Jeringas precargadas con 3 mg/3 ml: envases con 1 y 4.