BONVIVA® Comprimidos recubiertos
ROCHE
Tratamiento de osteoporosis posmenopáusica.
Bonviva es un bisfosfonato nitrogenado.
Composición.
Principio activo: Ácido ibandrónico, sal monosódica, monohidrato. Cada comprimido recubierto de 150 mg contiene 168,75 mg de ácido ibandrónico en forma de sal monosódica (monohidrato), equivalentes a 150 mg de ácido ibandrónico. Forma farmacéutica: Comprimidos recubiertos. Vía de administración: Oral. Lista de excipientes: Núcleo de los comprimidos: Lactosa monohidrato Ph. Eur./NF. Povidona Ph. Eur./USP. Celulosa microcristalina Ph. Eur./NF. Crospovidona Ph. Eur./NF. Ácido esteárico purificado Ph. Eur./NF. Sílice coloidal anhidra Ph. Eur./NF. Recubrimiento de los comprimidos: Hipromelosa Ph. Eur./USP. Dióxido de titanio Ph. Eur./USP. Talco Ph. Eur./USP. Macrogol 6000 Ph. Eur./NF.
Farmacología.
Propiedades farmacodinámicas: La acción farmacodinámica del ácido ibandrónico consiste en la inhibición de la resorción ósea. In vivo, el ácido ibandrónico previene la destrucción ósea inducida experimentalmente por el bloqueo de la función gonadal, retinoides, tumores o extractos tumorales. En ratas jóvenes (en crecimiento rápido) inhibe también la resorción ósea endógena, lo cual conduce a un aumento de la masa ósea en comparación con animales no tratados. Los modelos animales confirman que el ácido ibandrónico es un inhibidor muy potente de la actividad osteoclástica. En ratas en fase de crecimiento no se han observado indicios de alteración de la mineralización ni siquiera con dosis 5.000 veces superiores a la dosis necesaria para el tratamiento de la osteoporosis. La gran potencia y el amplio margen terapéutico del ácido ibandrónico permiten una mayor flexibilidad con las pautas posológicas, así como la posibilidad de tratamiento intermitente con dosis relativamente bajas e intervalos prolongados sin medicación. La administración prolongada diaria o intermitente (con largos intervalos sin medicación) a ratas, perros y monos se asoció con la formación de tejido óseo nuevo de calidad normal y/o un aumento de la resistencia mecánica incluso con dosis superiores a las previstas para el uso terapéutico, también dentro del intervalo de toxicidad. En la especie humana, la eficacia de la administración de ácido ibandrónico diaria o intermitente con un intervalo sin medicación de 9 - 10 semanas se confirmó en un estudio clínico (MF 4411) en el que Bonviva demostró su eficacia contra las fracturas. Tanto la administración diaria como intermitente (con un intervalo de 9 o 10 semanas sin medicación por trimestre) de Bonviva p.o. en mujeres posmenopáusicas produjo cambios bioquímicos indicativos de inhibición (dependiente de la dosis) de la resorción ósea, como disminución de los marcadores bioquímicos urinarios de la degradación del colágeno óseo (por ejemplo: desoxipiridinolina y telopéptidos C y N entrecruzados del colágeno de tipo I). Tras suspender el tratamiento, se apreciaba un regreso a los valores patológicos preterapéuticos de elevación de la resorción ósea característicos de la osteoporosis posmenopáusica. En un estudio de fase I de bioequivalencia en 72 mujeres posmenopáusicas tratadas con 150 mg p.o. cada 28 días hasta un total de cuatro dosis se observó inhibición del CTX en suero tras la primera dosis después de sólo 24 horas desde la administración (mediana de inhibición: 28%), registrándose la inhibición máxima (mediana: 69%) 6 días después. Tras la tercera y la cuarta dosis, la mediana de la inhibición máxima a los 6 días de la administración fue del 74%, descendiendo a una mediana de inhibición del 56% al cabo de 28 días de la cuarta dosis. Sin ninguna dosis más, se produce una pérdida de la supresión de marcadores bioquímicos de la resorción ósea. Mecanismo de acción: El ácido ibandrónico es un bisfosfonato muy potente, perteneciente al grupo de los bisfosfonatos nitrogenados, que actúa sobre el tejido óseo e inhibe de forma específica la actividad osteoclástica, sin interferir en el reclutamiento de los osteoclastos. La acción selectiva del ácido ibandrónico sobre el tejido óseo obedece a su elevada afinidad por la hidroxiapatita, que constituye la matriz mineral ósea. El ácido ibandrónico reduce la resorción ósea, sin afectar directamente a la formación del tejido óseo. En las mujeres posmenopáusicas disminuye el recambio óseo acelerado hacia niveles premenopáusicos, con lo que se produce un aumento neto progresivo de la masa ósea. La administración diaria o intermitente de ácido ibandrónico disminuye la resorción ósea, como reflejan la reducción de las concentraciones sérica y urinaria de los marcadores bioquímicos de recambio óseo, el aumento de la densidad mineral ósea (DMO) y el descenso de la incidencia de fracturas. Ensayos clínicos / Eficacia: Tratamiento de la osteoporosis posmenopáusica: En el estudio inicial sobre fracturas, de tres años, aleatorizado, de doble ciego y controlado con placebo (MF 4411), se demostró una reducción estadísticamente significativa y médicamente importante en la incidencia de nuevas fracturas vertebrales radiológicas (morfométricas) y clínicas. Bonviva se estudió en dosis orales de 2,5 mg diarios y 20 mg de forma intermitente (20 mg en días alternos hasta completar 12 dosis al comienzo de cada ciclo de 3 meses, separados por sendos períodos de reposo farmacológico de 9 - 10 semanas). Bonviva se administró 60 minutos antes del primer alimento (líquido o sólido) del día (período de ayuno postadministración). En el estudio participaron 2.946 mujeres de 55 a 80 años (en 2.928 de ellas era evaluable la eficacia), con 5 años como mínimo de posmenopausia, una DMO de 2 - 5 desviaciones típicas (DE) por debajo de la media premenopáusica (puntuación T) en al menos una vértebra lumbar (L1 - L4) y antecedentes de una a cuatro fracturas vertebrales. Todas las pacientes recibieron diariamente 500 mg de calcio y 400 UI de vitamina D. En ambos grupos de tratamiento con Bonviva se demostró una reducción estadísticamente significativa y médicamente importante de la incidencia de nuevas fracturas vertebrales. Con la pauta de 2,5 mg diarios, la incidencia de nuevas fracturas vertebrales radiográficas disminuyó en un 62% a lo largo de los tres años del estudio, mientras que las fracturas vertebrales clínicas disminuyeron en un 49%. Este potente efecto en las fracturas vertebrales se reflejó asimismo en una disminución estadísticamente significativa de la pérdida de estatura en comparación con placebo. Este efecto protector contra las fracturas fue constante durante todo el estudio, y no se apreciaron indicios de desvanecimiento del efecto con el paso del tiempo. Aunque este estudio clínico no estaba específicamente diseñado para demostrar la eficacia del ácido ibandrónico en las fracturas extravertebrales, en un subgrupo de pacientes con alto riesgo de fractura pudo comprobarse una reducción del riesgo relativo de fracturas extravertebrales (DMO en el cuello femoral: puntuación T < - 3,0 DE) de magnitud similar (69%) a la observada para las fracturas vertebrales. La observación de eficacia en las fracturas extravertebrales de subgrupos de alto riesgo concuerda bien con los resultados de los estudios clínicos realizados con otros bisfosfonatos. Con la pauta de administración diaria, la DMO lumbar aumentó en un 5,3% con respecto al placebo al cabo de los tres años de estudio; en comparación con los valores iniciales, el aumento fue del 6,5%. Los marcadores bioquímicos de recambio óseo (como CTX urinario y osteocalcina sérica) pusieron de manifiesto el patrón previsto de inhibición hasta niveles premenopáusicos, con valores máximos de inhibición al cabo de 3 - 6 meses. Tan sólo un mes después de iniciado el tratamiento con Bonviva se observó ya una reducción del 50% (con la pauta de 2,5 mg diarios) y del 78% (con la pauta de 20 mg de forma intermitente) en los marcadores bioquímicos de resorción ósea, en ambos casos de importancia clínica. La disminución de los marcadores bioquímicos de resorción ósea fue ya evidente en la primera semana de tratamiento. Bonviva en una dosis mensual de 150 mg: Densidad mineral ósea: Bonviva en una dosis mensual de 150 mg demostró ser al menos tan eficaz como en una dosis diaria de 2,5 mg con una DMO creciente en un estudio multicéntrico de dos años, de doble ciego (BM 16549), realizado en mujeres posmenopáusicas con osteoporosis (T de DMO inicial en la columna lumbar: < -2,5 DE). Este efecto se demostró tanto en el análisis principal después de un año como en el análisis confirmatorio después de dos años (tabla 3).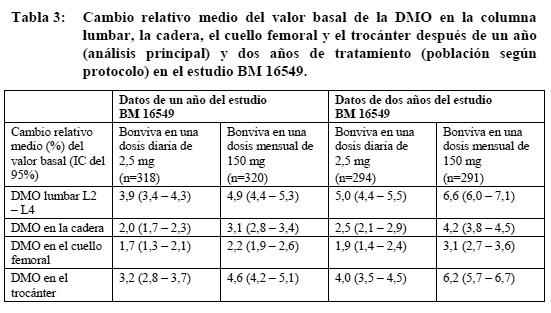
En un análisis prospectivo programado, Bonviva en una dosis mensual de 150 mg fue superior a Bonviva en una dosis diaria de 2,5 mg para aumentar la DMO lumbar después de un año (p=0,002) y dos años (p=,001). Al cabo de un año (análisis principal), el 91,3% (p=0,005) de las pacientes tratadas con 150 mg de Bonviva una vez al mes presentaban un aumento de la DMO lumbar igual o superior al valor basal (respondedoras de la DMO), frente al 84,0% de los las pacientes que habían recibido 2,5 mg de Bonviva al día. Al cabo de dos años eran respondedoras el 93,5% (p=0,004) y el 86,4% de las pacientes tratadas con 150 mg de Bonviva una vez al mes o 2,5 mg de Bonviva al día. Por lo que respecta a la DMO en la cadera, el 90,0% (p < 0,001) de las pacientes tratadas con 150 mg de Bonviva una vez al mes y el 76,5% de las que recibieron 2,5 mg de Bonviva al día presentaban después de un año un aumento de la DMO en la cadera igual o superior al valor basal. Al cabo de dos años, el 93,4% (p < 0,001) de las pacientes tratadas con 150 mg de Bonviva una vez al mes y el 78,4% de las que recibieron 2,5 mg de Bonviva al día presentaban un aumento de la DMO en la cadera igual o superior al valor basal. Si se aplica un criterio más estricto, la combinación de la DMO en la columna lumbar y la cadera arroja un 83,9% (p < 0,001) y un 65,7% de pacientes respondedoras con 150 mg de Bonviva una vez al mes y 2,5 mg de Bonviva al día, respectivamente. Al cabo de dos años cumplían este criterio el 87,1% (p=0,001) y el 70,5% de las pacientes tratadas con 150 mg de Bonviva una vez al mes y 2,5 mg de Bonviva al día, respectivamente. Marcadores bioquímicos del recambio óseo: En todos los análisis, es decir, en los meses 3, 6, 12 y 24, se midieron reducciones clínicamente importantes de la concentración sérica de CTX. Después de un año (análisis principal), la mediana del cambio relativo del valor basal era del -76% con 150 mg de Bonviva una vez al mes y del -67% con 2,5 mg de Bonviva al día. Al cabo de dos años, la mediana del cambio relativo era del -68% y el -62% en los grupos con 150 mg una vez al mes y 2,5 mg al día de Bonviva, respectivamente. Después de un año habían alcanzado el criterio de respondedoras (descenso ≥ 50% del valor basal) el 83,5% (p=0,006) de las pacientes tratadas con 150 mg de Bonviva una vez al mes y el 73,9% de las tratadas con 2,5 mg de Bonviva al día. Al cabo de dos años cumplían este criterio el 78,7% (p=0,002) y el 65,6% de las pacientes de los grupos con 150 mg una vez al mes y 2,5 mg al día de Bonviva, respectivamente. Considerando los resultados del estudio BM 16549, se espera que Bonviva en una dosis mensual de 150 mg sea al menos tan eficaz como en una dosis diaria de 2,5 mg en la prevención de las fracturas. Propiedades farmacocinéticas: Los efectos farmacológicos del ácido ibandrónico no guardan una relación directa con sus concentraciones plasmáticas. Así se ha observado en estudios realizados tanto con animales como en el ser humano, en los cuales se demostró una eficacia equivalente del ácido ibandrónico en administración diaria o intermitente, con un intervalo de reposo farmacológico de varias semanas (como mínimo 6 semanas en las ratas, 11 semanas en los perros, 30 días en los monos, 9,5 semanas en el ser humano), siempre y cuando se administrara la misma dosis total durante este periodo. Absorción: Tras su administración, el ácido ibandrónico se absorbe rápidamente en el tubo digestivo superior. La concentración plasmática aumenta proporcionalmente hasta la dosis de 50 mg por vía oral, siendo su aumento sobreproporcional por encima de esta dosis. Las concentraciones plasmáticas máximas observadas se alcanzaron entre 0,5 y 2 h (mediana: 1 h) después de la dosis en ayunas. La biodisponibilidad absoluta era del orden del 0,6%. El grado de absorción disminuye cuando el ácido ibandrónico se administra con alimentos sólidos o líquidos (excepto el agua natural). Cuando el ácido ibandrónico se administra con un desayuno normal, su biodisponibilidad disminuye en torno al 90% con respecto a la alcanzada en ayunas. No se aprecia ninguna disminución importante de la biodisponibilidad cuando el ácido ibandrónico se administra una hora antes de una comida. Tanto la biodisponibilidad como el aumento de la DMO disminuyen cuando se administra un alimento sólido o líquido menos de una hora después de la dosis de Bonviva. Distribución: Tras la exposición sistémica inicial, el ácido ibandrónico se une rápidamente al hueso o se excreta con la orina. En el ser humano, el volumen terminal aparente de distribución es de 90 l como mínimo, y la cantidad de fármaco que llega al tejido óseo se calcula en un 40 - 50% de la dosis circulante. La fijación a las proteínas plasmáticas es baja (aproximadamente del 85% en concentraciones terapéuticas), de modo que el riesgo de interacciones farmacológicas por desplazamiento es pequeño. Metabolismo: No hay indicios de que el ácido ibandrónico se metabolice en los animales o en el ser humano. Eliminación: La fracción absorbida de ácido ibandrónico desaparece de la circulación por absorción ósea (40 - 50%), y el resto se elimina de forma inalterada por los riñones. La fracción no absorbida de ácido ibandrónico se elimina inalterada con las heces. Se ha observado un amplio intervalo de valores de semivida aparente de eliminación según la dosis y la sensibilidad del análisis utilizado, pero la semivida terminal aparente suele hallarse entre 10 y 72 horas. La concentración plasmática inicial desciende rápidamente hasta un 10% de los valores máximos en el espacio de 3 horas tras la administración intravenosa (i.v.) y 8 horas tras la administración oral. El aclaramiento total del ácido ibandrónico es bajo, con valores medios que oscilan entre 84 y 160 ml/min. El aclaramiento renal (aproximadamente 60 ml/min en las mujeres posmenopáusicas sanas) representa el 50 - 60% del aclaramiento total y está relacionado con el aclaramiento de la creatinina. La diferencia entre el aclaramiento renal y el aclaramiento total aparente se atribuye a la captación ósea. Farmacocinética en poblaciones especiales: Sexo: La biodisponibilidad y la farmacocinética del ácido ibandrónico son similares en los hombres y las mujeres. Raza: No hay indicios de diferencias interétnicas de importancia clínica entre la población asiática y la caucasiana en cuanto a disposición del ácido ibandrónico. Son pocos los datos disponibles sobre pacientes de raza negra. Insuficiencia renal: El aclaramiento renal del ácido ibandrónico guarda una relación lineal con el aclaramiento de la creatinina (Acr) en pacientes con diversos grados de insuficiencia renal. No es necesario ajustar la dosis en pacientes con insuficiencia renal leve o moderada (Acr ≥ 30 ml/min), como se ha puesto de manifiesto en el estudio BM 16549, en el que la mayoría de las pacientes pertenecía a una u otra categoría. Sujetos con insuficiencia renal grave (Acr < 30 ml/min) tratados con 10 mg diarios de ácido ibandrónico p.o. durante 21 días presentaron concentraciones plasmáticas 2 - 3 veces mayores que los sujetos con normofunción renal (aclaramiento total = 129 ml/min). El aclaramiento total del ácido ibandrónico se redujo a 44 ml/min en los sujetos con insuficiencia renal grave. Tras la administración de 0,5 mg por vía i.v., las cifras de aclaramiento disminuyeron en un 67% (aclaramiento total), un 77% (aclaramiento renal) y un 50% (aclaramiento extrarrenal) en los sujetos con insuficiencia renal grave. El consiguiente aumento de la exposición no comportaba, sin embargo, una reducción de la tolerabilidad. Insuficiencia hepática: No existen datos farmacocinéticos disponibles sobre el ácido ibandrónico en pacientes con insuficiencia hepática. El hígado no desempeña ninguna función importante en el aclaramiento del ácido ibandrónico, que no se metaboliza, sino que desaparece de la sangre por excreción renal y captación ósea. Por lo tanto, no es necesario ajustar la dosis en los pacientes con insuficiencia hepática. Además, dado que la fijación del ácido ibandrónico a las proteínas plasmáticas es baja en concentraciones terapéuticas (85%), parece poco probable que la hipoproteinemia de las hepatopatías graves pueda dar lugar a aumentos clínicamente importantes de la concentración plasmática de fármaco libre. Ancianos: En un análisis estadístico multifactorial, la edad no fue un factor independiente para ninguna de las variables farmacocinéticas estudiadas. Dado que la función renal disminuye con la edad, éste es el único factor que debe tomarse en consideración (ver Insuficiencia renal). Niños: No existen datos sobre el uso de Bonviva en niños y adolescentes menores de 18 años. Datos preclínicos sobre seguridad: En los animales de experimentación, efectos tóxicos sólo se observaron con exposiciones consideradas suficientemente superiores a la exposición humana máxima, lo que era indicativo de poca importancia para el uso clínico. Carcinogenicidad: No se ha observado ningún indicio de potencial carcinógeno. Mutagenicidad: No se ha observado ningún indicio de potencial genotóxico.
Indicaciones.
Bonviva en comprimidos de 150 mg está indicado para el tratamiento y la prevención de la osteoporosis posmenopáusica.
Dosificación.
La dosis recomendada de Bonviva para el tratamiento es de un comprimido recubierto de 150 mg una vez al mes. Los comprimidos deben tomarse preferiblemente en el mismo día de cada mes. Bonviva debe administrarse 60 minutos antes del primer alimento sólido o líquido del día (sin contar el agua) (ver Interacciones; Interacciones con los alimentos) o cualquier otro medicamento oral (incluidos los suplementos de calcio). Los comprimidos deben tragarse enteros acompañados de un vaso de agua natural (180 - 240 ml), estando la paciente o en posición erecta, ya sea sentada o de pie. Las pacientes no deben recostarse hasta 60 minutos después de haber ingerido Bonviva. El agua natural es la única bebida que puede tomarse con Bonviva. Adviértase que ciertas aguas minerales pueden tener concentraciones elevadas de calcio y, por lo tanto, no deben utilizarse. Los comprimidos no deben masticarse ni disolverse en la boca a causa del riesgo de úlceras bucofaríngeas. Las pacientes deben recibir suplementos de calcio o vitamina D cuando el consumo de estos nutrientes con la alimentación sea insuficiente. Se debe instruir a las pacientes para que, si olvidan una dosis mensual, tomen un comprimido de Bonviva de 150 mg en la mañana siguiente de acordarse, salvo que el número de días hasta la siguiente dosis prevista (programada) sea de 7 o menor. La dosis mensual siguiente deben tomarla de nuevo en el día previsto originalmente. Si el número de días hasta la siguiente dosis prevista es de 7 o menor, las pacientes deben esperar hasta la dosis siguiente y seguir después tomando un comprimido al mes según el plan original. Las pacientes no deben tomar dos comprimidos de 150 mg en una misma semana. Pautas posológicas especiales: Insuficiencia hepática: No es necesario ajustar la dosis (ver Farmacocinética en poblaciones especiales). Insuficiencia renal: No es necesario ajustar la dosis en las pacientes con insuficiencia renal leve o moderada, con cifras de aclaramiento de la creatinina ≥ 30 ml/min. En caso de aclaramiento de la creatinina < 30 ml/min, (insuficiencia renal severa), el uso de este medicamento no está recomendado, debido a que la experiencia clínica es limitada. Ancianos: No es necesario ajustar la dosis. Niños: No se han determinado la seguridad y la eficacia en pacientes menores de 18 años.
Contraindicaciones.
Bonviva está contraindicado en pacientes con antecedentes de alergia al ácido ibandrónico o a alguno de los excipientes. Bonviva está contraindicado en pacientes con hipocalcemia no corregida. Como con todos los bisfosfonatos indicados para el tratamiento de la osteoporosis, la hipocalcemia preexistente ha de corregirse antes de comenzar la administración de Bonviva. Bonviva, como varios bisfosfonatos, está contraindicado en pacientes con anomalías del esófago que retarden el vaciado esofágico, como una constricción o acalasia (ver Advertencias). Bonviva está contraindicado asimismo en pacientes incapacitadas para permanecer levantadas o sentadas en posición erecta durante un mínimo de 60 minutos (ver Dosificación y Advertencias).
Reacciones adversas.
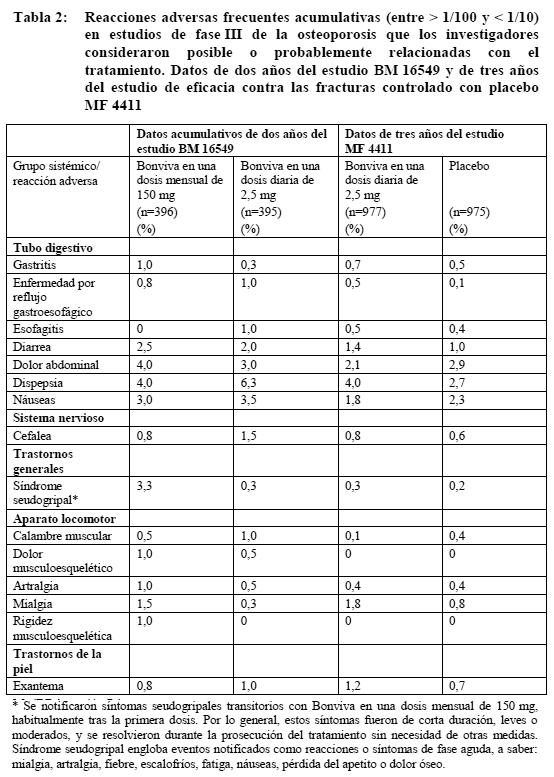
Ensayos clínicos: Tratamiento de la osteoporosis posmenopáusica: Con una dosis mensual: En un estudio de dos años en mujeres posmenopáusicas con osteoporosis (BM 16549), la seguridad global de Bonviva en una dosis mensual de 150 mg y una dosis diaria de 2,5 mg fue similar. La proporción global de pacientes con reacciones adversas, es decir, acontecimientos adversos posible o probablemente relacionados con la medicación en estudio, fue del 22,7% y el 25,0% con Bonviva en una dosis mensual de 150 mg y una dosis diaria de 2,5 mg al cabo de uno y dos años, respectivamente. La mayoría de las reacciones adversas fueron de intensidad leve o moderada, y en la mayor parte de los casos no obligaron a suspender el tratamiento. Las tablas 1 y 2 recogen las reacciones adversas observadas en más del 1% de las pacientes tratadas con Bonviva en una dosis mensual de 150 mg o una dosis diaria de 2,5 mg en el estudio BM 16549, así como en las pacientes tratadas con Bonviva en una dosis diaria de 2,5 mg en el estudio MF 4411. Las tablas muestran las reacciones adversas de los dos estudios que tuvieron una incidencia mayor que la registrada en las pacientes que habían recibido placebo en el estudio MF 4411. Dentro de cada grupo de frecuencia, los efectos adversos se presentan en orden decreciente de gravedad. Los datos de un año del estudio BM 16549 se presentan en la tabla 1, y los datos acumulativos de dos años del estudio BM 16549, en la tabla 2.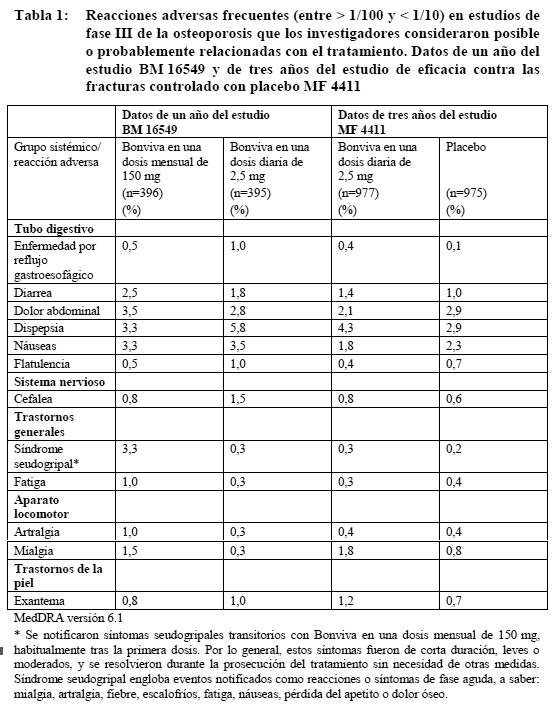
Reacciones adversas con una frecuencia del 1% como máximo. En la lista siguiente se muestran las reacciones adversas (consideradas por los investigadores como posible o probablemente relacionadas con el tratamiento) notificadas en el estudio MF 4411 cuya frecuencia con 2,5 mg de Bonviva al día era mayor que con placebo y las notificadas en el estudio BM 16549 cuya frecuencia con 150 mg de Bonviva una vez al mes era mayor que con 2,5 mg de Bonviva al día. Dentro de cada grupo de frecuencia, los efectos adversos se presentan en orden decreciente de gravedad. Poco frecuente (entre 1/100 y 1/1.000): Trastornos gastrointestinales: gastritis, esofagitis incluidas ulceración esofágica o constricciones, vómitos, disfagia. Trastornos del sistema nervioso: mareos. Trastornos musculoesqueléticos y del tejido conjuntivo: dolor de espalda. Raras veces (entre 1/1.000 y 1/10.000): Trastornos gastrointestinales: duodenitis. Trastornos del sistema inmunitario: reacciones de hipersensibilidad. Trastornos de la piel y el tejido subcutáneo: angioedema, edema facial, urticaria. En el estudio de tratamiento con una dosis al mes se incluyó a pacientes con antecedentes de enfermedad gastrointestinal, incluida úlcera péptica sin hemorragia reciente u hospitalización, y a pacientes con dispepsia o reflujo controlados con medicación. En estas pacientes, entre el régimen de una dosis mensual de 150 mg y el de una dosis diaria de 2,5 mg no hubo diferencias en la incidencia de reacciones en el tubo digestivo superior. Alteraciones analíticas: En el estudio fundamental de tres años con Bonviva en una dosis diaria de 2,5 mg (MF 4411) no hubo diferencias con el grupo de placebo en cuanto a alteraciones analíticas indicativas de disfunción hepática o renal, trastornos hematológicos, hipocalcemia e hipofosfatemia. De igual modo, no se registraron diferencias entre los grupos en el estudio BM 16549 después de uno y dos años. Experiencia tras la comercialización: Trastornos musculoesqueléticos y del tejido conjuntivo: En pacientes tratadas con ácido ibandrónico se ha descrito osteonecrosis de la mandíbula en muy raras ocasiones (ver Advertencias). Trastornos oculares: Con bisfosfonatos, el ácido ibandrónico incluido, se han descrito episodios de inflamación ocular, como uveítis, epiescleritis y escleritis. En algunos casos, la resolución de estos episodios no se produjo hasta la retirada del bisfosfonato. Trastornos del sistema inmunitario: Se han descrito casos de reacción anafiláctica/shock, incluidos algunos de desenlace fatal, en pacientes tratados con ácido ibandrónico.
Advertencias.
Antes de iniciar el tratamiento con Bonviva es preciso corregir la hipocalcemia y otros trastornos del metabolismo óseo y mineral. En todas las pacientes es importante que el consumo de calcio y vitamina D sea suficiente. Los bisfosfonatos administrados p.o. pueden causar irritación local de la mucosa del tubo digestivo superior. Dada la posibilidad de este efecto irritante y de empeoramiento de la enfermedad subyacente, se procederá con especial precaución cuando Bonviva se administre a pacientes con trastornos activos del tubo digestivo superior (por ejemplo: esófago de Barrett, disfagia, otras dolencias esofágicas, gastritis, duodenitis o úlcera). En pacientes tratadas con bisfosfonatos orales se han descrito acontecimientos adversos como esofagitis, úlceras y erosiones esofágicas, que en algunos casos fueron graves y requirieron la hospitalización; en raras ocasiones se acompañaron de hemorragia o les siguieron constricción o perforación esofágica. El riesgo de graves acontecimientos adversos esofágicos parece ser mayor en las pacientes que no cumplen las pautas posológicas y/o siguen tomando bisfosfonatos orales después de presentarse síntomas que sugieren irritación esofágica. Las pacientes deben prestar especial atención a las pautas posológicas y cumplirlas (ver Dosificación). Los médicos deben estar atentos a cualquier signo o síntoma indicativo de una posible reacción esofágica y advertir, además, a las pacientes que suspendan el tratamiento con Bonviva y soliciten asistencia médica si presentan disfagia, odinofagia, dolor retroesternal o pirosis de nueva aparición o empeorada. Aunque no se observó un aumento del riesgo en los estudios clínicos controlados, ha habido notificaciones tras la comercialización de úlceras de estómago o duodeno tras la administración de bisfosfonatos orales, en algunos casos graves y con complicaciones. Dado que los AINE y los bisfosfonatos se asocian a irritación gastrointestinal, deben extremarse las precauciones en caso de coadministración con Bonviva. Se ha descrito osteonecrosis de la mandíbula (ONM) en pacientes tratadas con bisfosfonatos. La mayoría eran pacientes con cáncer sometidas a procedimientos dentales, pero en algunos casos se trataba de pacientes con osteoporosis posmenopáusica u otros diagnósticos. Entre los factores de riesgo conocidos se hallan el diagnóstico de cáncer, tratamientos concomitantes (por ejemplo: quimioterapia, radioterapia, corticosteroides) y trastornos de comorbilidad (por ejemplo: anemia, coagulopatía, infección, enfermedad dental preexistente). Los casos notificados han sido en su mayor parte de pacientes tratadas con bisfosfonatos por vía i.v., pero en algunas la vía de administración fue la oral. En las pacientes que experimenten ONM durante el tratamiento con bisfosfonatos, la cirugía dental puede empeorar su estado. Para las pacientes que requieran un procedimiento dental, no existen datos que indiquen si la suspensión del tratamiento con bisfosfonatos reduce el riesgo de ONM. El juicio clínico del médico debe guiarle en su actuación terapéutica con cada paciente de acuerdo con la valoración individual del balance de riesgos y beneficios. Efectos sobre la capacidad para conducir y utilizar máquinas: No se han llevado a cabo estudios sobre los efectos en la capacidad de conducir vehículos y utilizar maquinaria.
Interacciones.
Interacciones con los alimentos: Los productos que contienen calcio y otros cationes multivalentes (por ejemplo: aluminio, magnesio, hierro), como la leche y otros alimentos, pueden interferir con la absorción de Bonviva, según se ha observado en los estudios con animales. Por lo tanto, hasta la ingestión de tales productos o alimentos deben transcurrir 60 minutos desde la última administración oral. Interacciones farmacológicas: Es probable que los suplementos de calcio, los antiácidos y algunos medicamentos orales que contienen cationes multivalentes (por ejemplo: aluminio, magnesio, hierro) puedan interferir con la absorción de Bonviva. Por lo tanto, las pacientes deben esperar 60 minutos después de haber ingerido Bonviva antes de tomar otros medicamentos orales. En los estudios de interacciones farmacocinéticas en mujeres posmenopáusicas se ha demostrado la ausencia de potencial de interacción con el tamoxifeno y la estrogenoterapia de restitución (estrógenos). No se han observado interacciones con la administración simultánea de Bondronat y melfalano/prednisolona en pacientes con mieloma múltiple. En voluntarios sanos de sexo masculino y mujeres posmenopáusicas, la ranitidina i.v. aumentó la biodisponibilidad del ácido ibandrónico en torno al 20%, probablemente como resultado de la disminución de la acidez gástrica. Ahora bien, dado que este aumento se halla dentro de los valores normales de la biodisponibilidad del ácido ibandrónico, no se considera necesario ajustar la dosis de Bonviva cuando se asocie a los antihistamínicos H2 u otros fármacos que elevan el pH gástrico. En relación con la disposición, no es probable ninguna interacción de importancia clínica, puesto que el ácido ibandrónico no inhibe las principales enzimas hepáticas humanas del citocromo P450 y tampoco induce este sistema enzimático en las ratas. Además, la fijación a las proteínas plasmáticas es baja en concentraciones terapéuticas, por lo que no parece probable que el ácido ibandrónico desplace a otros fármacos. El ácido ibandrónico se elimina exclusivamente por excreción renal y no sufre ningún tipo de biotransformación. Parece que la vía de excreción no incluye los conocidos sistemas de transporte ácido o básico implicados en la excreción de otros fármacos. En un estudio de un año en mujeres posmenopáusicas con osteoporosis (BM 16549), la incidencia de reacciones en el tubo digestivo superior en las pacientes que tomaban simultáneamente ácido acetilsalicílico (Aspirina) o AINE fue similar a la registrada en las pacientes tratadas con Bonviva en una dosis de 2,5 mg diarios o de 150 mg una vez al mes. De las más de 1.500 pacientes participantes en el estudio BM 16549, en el que se comparaba la administración mensual con la diaria de ácido ibandrónico, el 14% de ellas utilizaba antihistamínicos (antagonistas de los receptores H2) o inhibidores de la bomba de protones. En estas pacientes, la incidencia de reacciones en el tubo digestivo superior entre las tratadas con Bonviva en una dosis mensual de 150 mg fue similar a la observada entre las que recibieron 2,5 mg diarios de Bonviva. Uso en poblaciones especiales: Embarazo: Bonviva no debe utilizarse durante el embarazo. No se observaron signos de teratogenia ni toxicidad fetal directa en las ratas y conejos tratados con ácido ibandrónico p.o. diariamente, ni tampoco efectos adversos sobre el desarrollo de la descendencia (primera generación, F1) de las ratas. Los efectos adversos del ácido ibandrónico en los estudios de toxicidad en la reproducción de ratas fueron idénticos a los observados con los bisfosfonatos como grupo terapéutico. Entre ellos se cuentan la disminución del número de sitios de implantación, la interferencia con el parto natural (distocia) y el aumento de variaciones viscerales (síndrome ureteropielorrenal). No se han realizado estudios específicos con el régimen de administración mensual. No existe experiencia clínica sobre el uso de Bonviva en mujeres embarazadas. Lactancia: Bonviva no debe utilizarse durante la lactancia. En las ratas lactantes tratadas con 0,08 mg/kg/día de ácido ibandrónico por vía i.v., la concentración máxima de ácido ibandrónico en la leche materna fue de 8,1 ng/ml y se registró en las 2 primeras horas tras la administración i.v. Al cabo de 24 h, la concentración en la leche fue similar a la concentración en el plasma. Correspondió a un 5%, aproximadamente, de la concentración medida a las 2 h de administrada la dosis. Se ignora si Bonviva pasa a la leche materna humana. Uso en pediatría: Ver Farmacocinética en poblaciones especiales, Niños. Uso en geriatría: Ver Farmacocinética en poblaciones especiales, Ancianos. Insuficiencia renal: Ver Farmacocinética en poblaciones especiales, Insuficiencia renal. Insuficiencia hepática: Ver Farmacocinética en poblaciones especiales, Insuficiencia hepática.
Conservación.
No debe conservarse a más de 30°C. Instrucciones especiales de uso, manipulación y eliminación: Este medicamento sólo deberá utilizarse hasta la fecha de caducidad, indicada con "VEN" en el envase. Eliminación de medicamentos no utilizados/caducados: La emisión de productos farmacéuticos al medio ambiente debe reducirse a un mínimo. Los medicamentos no deben eliminarse a través de las aguas residuales, y su eliminación con los residuos domésticos también debe evitarse. Utilice los sistemas de recogida establecidos si los hay en su localidad.
Sobredosificación.
No hay información específica sobre el tratamiento de una sobredosis de Bonviva. No obstante, es de esperar que las sobredosis puedan causar efectos secundarios de tipo digestivo alto, como malestar gástrico, pirosis, esofagitis, gastritis o úlcera gástrica. Puede administrarse leche o antiácidos para fijar Bonviva. Dado el riesgo de irritación esofágica, se desaconseja la provocación del vómito y se aconseja mantener a la paciente en posición totalmente erecta.
Presentación.
Comprimidos recubiertos de 150 mg 1, 3.