BOSULIF
PFIZER
Antineoplásico.
Composición.
Comprimidos de bosutinib de 100 mg: cada comprimido contiene bosutinib monohidrato equivalente a 100 mg de bosutinib. Comprimidos de bosutinib de 400 mg: cada comprimido contiene bosutinib monohidrato equivalente a 400 mg de bosutinib. Comprimidos de bosutinib de 500 mg: cada comprimido contiene bosutinib monohidrato equivalente a 500 mg de bosutinib.
Indicaciones.
Bosutinib está indicado para el tratamiento de pacientes adultos con leucemia mielógena crónica con cromosoma Philadelphia positivo (LMC Ph+) en fase crónica (FC) recientemente diagnosticada. El bosutinib está indicado para el tratamiento de pacientes adultos con leucemia mieloide crónica con cromosoma Philadelphia positivo (LMC Ph+) en fase crónica, acelerada o blástica con resistencia o intolerancia al tratamiento previo.
Dosificación.
El tratamiento debe iniciarlo un médico con experiencia en el diagnóstico y el tratamiento de pacientes con LMC. Posología: LMC Ph+ en FC recién diagnosticada. La dosis recomendada es de 400 mg de bosutinib una vez al día. LMC Ph+ en FC, FA o FB con resistencia o intolerancia al tratamiento previo. La dosis recomendada es de 500 mg de bosutinib una vez al día. En los ensayos clínicos para ambas indicaciones, se mantuvo el tratamiento con bosutinib hasta la progresión de la enfermedad o la intolerancia al tratamiento. Ajustes de dosis: En el estudio clínico de fase 1/2 en pacientes con LMC que eran resistentes o intolerantes al tratamiento previo, se permitieron aumentos de la dosis de 500 mg a 600 mg una vez al día con alimentos en pacientes que no mostraron una respuesta hematológica completa (RHC) transcurridas 8 semanas o una respuesta citogenética completa (RC y C) transcurridas 12 semanas y que no tuvieran acontecimientos adversos posiblemente relacionados con el producto en investigación de grado 3 o superior. Sin embargo, en el estudio de fase 3 en pacientes con LMC en FC recientemente diagnosticada tratados con 400 mg de bosutinib se permitieron aumentos de la dosis en incrementos de 100 mg hasta un máximo de 600 mg una vez al día con alimentos si el paciente no mostró transcritos de BCR-ABL ≤10% en el mes 3, no tuvo una reacción adversa de grado 3 o 4 en el momento del aumento, y todas las toxicidades no hematológicas de grado 2 remitieron al menos hasta el grado 1. En el estudio clínico de fase 1/2 en pacientes con LMC resistentes o intolerantes al tratamiento previo que comenzaron el tratamiento con ≤500 mg, 93 (93/558; 16,7%) pacientes tuvieron aumentos de la dosis hasta 600 mg al día. En el estudio de fase 3 en pacientes con LMC en FC recién diagnosticada que iniciaron el tratamiento con bosutinib a 400 mg, un total de 46 pacientes (17,2%) recibieron aumentos de la dosis hasta 500 mg. Además, el 5,6% de los pacientes en el grupo de tratamiento con bosutinib tuvieron aumentos de la dosis adicionales hasta 600 mg. No se han estudiado dosis superiores a 600 mg/día y por lo tanto, no deben administrarse. Ajustes de dosis en función de las reacciones adversas Reacciones adversas no hematológicas. Si aparece toxicidad no hematológica moderada o grave, clínicamente significativa, debe interrumpirse el tratamiento con bosutinib, y una vez resuelta la toxicidad se puede reanudar el tratamiento a una dosis reducida en 100 mg tomada una vez al día. Si resulta adecuado desde el punto de vista clínico, puede plantearse ir aumentando la dosis hasta alcanzar la dosis previa a la reducción de dosis tomada una vez al día. Se han utilizado dosis inferiores a 300 mg/día en pacientes; sin embargo, no se ha establecido la eficacia. Transaminasas hepáticas elevadas: si aparecen aumentos en las transaminasas hepáticas por encima de 5 veces el límite superior de la normalidad (LSN), debe interrumpirse el tratamiento con bosutinib hasta que se recupere una concentración ≤2,5 veces el LSN, tras lo que puede reanudarse el tratamiento a dosis de 400 mg una vez al día. Si la recuperación tarda más de 4 semanas, debe considerarse el cese del tratamiento con bosutinib. Si aparecen elevaciones de las transaminasas ≥3 veces el LSN junto con incrementos de la bilirrubina > 2 veces el LSN y en la fosfatasa alcalina < 2 veces el LSN, se debe interrumpir el tratamiento con bosutinib. Diarrea: en caso de diarrea de grado 3-4 conforme a los Criterios Comunes de Terminología para Reacciones Adversas (CTCAE, por sus siglas en inglés) del NCI, el tratamiento con bosutinib debe interrumpirse, y tras la recuperación hasta grado ≤1, puede reanudarse el tratamiento a dosis de 400 mg una vez al día. Reacciones adversas hematológicas: Se recomienda reducir la dosis en caso de neutropenia o trombocitopenia grave o persistente, tal y como se describe en la Tabla 1:
Poblaciones especiales: Pacientes de edad avanzada (≥65 años): En los pacientes de edad avanzada no es necesaria una recomendación específica sobre la dosis. Dado que la información en pacientes de edad avanzada es limitada, se debe tener precaución en estos pacientes. Insuficiencia renal: En los estudios de LMC se excluyó a los pacientes con creatinina sérica > 1,5 veces el LSN. Durante los estudios se observó un aumento en la exposición (ABC [área bajo la curva]) en los pacientes con insuficiencia renal moderada y grave. LMC Ph+ en FC recién diagnosticada: En pacientes con insuficiencia renal moderada (aclaramiento de creatinina [ClCr] 30 a 50 ml/min, calculado según la fórmula Cockcroft-Gault), la dosis recomendada de bosutinib es de 300 mg al día con alimentos. En pacientes con insuficiencia renal grave (ClCr < 30 ml/min, calculado según la fórmula Cockcroft-Gault), la dosis recomendada de bosutinib es de 200 mg al día con alimentos. Se puede considerar el aumento de la dosis a 400 mg una vez al día con alimentos para pacientes con insuficiencia renal moderada o a 300 mg una vez al día para pacientes con insuficiencia renal grave si no experimentan reacciones adversas graves o moderadas persistentes y si no alcanzan una respuesta hematológica, citogenética o molecular adecuada. LMC Ph+ en FC, FA o FB con resistencia o intolerancia al tratamiento previo: En pacientes con insuficiencia renal moderada (ClCr 30 a 50 ml/min, calculado según la fórmula Cockcroft-Gault), la dosis recomendada de bosutinib es de 400 mg al día. En pacientes con insuficiencia renal grave (ClCr < 30 ml/min, calculado según la fórmula Cockcroft- Gault), la dosis recomendada de bosutinib es de 300 mg al día. Se puede considerar un aumento de la dosis a 500 mg una vez al día en pacientes con insuficiencia renal moderada o a 400 mg una vez al día en pacientes con insuficiencia renal grave en los casos en los que no se hayan experimentado reacciones adversas graves o moderadas persistentes y si no alcanzan una respuesta hematológica, citogenética o molecular adecuada. Trastornos cardiacos: En los estudios clínicos se excluyó a los pacientes con cardiopatías no controladas o importantes (por ejemplo, infarto de miocardio reciente, insuficiencia cardiaca congestiva o angina inestable). En el caso de los pacientes con trastornos cardiacos importantes debe procederse con precaución. Trastorno gastrointestinal reciente o en curso, clínicamente significativo: En los estudios clínicos, se excluyó a los pacientes con trastorno gastrointestinal reciente o en curso clínicamente significativo (por ejemplo, vómitos y/o diarrea graves). En el caso de los pacientes con trastorno gastrointestinal reciente o en curso, clínicamente significativo, debe procederse con precaución. Población pediátrica: No se ha establecido la seguridad y eficacia de bosutinib en niños menores de 18 años de edad. No se dispone de datos. Forma de administración: Bosulif se debe tomar por vía oral una vez al día junto con alimentos. Si el paciente olvida tomar una dosis durante más de 12 horas, no se le debe administrar una dosis adicional. El paciente debe tomar la dosis prescrita habitual al día siguiente.
Contraindicaciones.
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos. Insuficiencia hepática.
Reacciones adversas.
Resumen del perfil de seguridad: Un total de 1272 pacientes con leucemia recibieron al menos una dosis de bosutinib como agente único. La mediana de la duración del tratamiento fue de 13,8 meses (intervalo: de 0,03 a 123,3 meses). Dichos pacientes tenían LMC en fase crónica recién diagnosticada, o bien LMC en fase crónica, acelerada o blástica resistente o intolerante a tratamientos anteriores, o bien tenían leucemia linfoblástica aguda (LLA) Ph+. De dichos pacientes, 268 (dosis inicial de 400 mg) y 248 (dosis inicial de 500 mg) procedían de los 2 estudios de fase 3 realizado en pacientes con LMC no tratados previamente, 570 y 63 pacientes procedían de 2 estudios de fase 1/2 realizados en pacientes con leucemias Ph+ tratadas previamente y 123 pacientes procedían de un estudio de fase 4 realizado en pacientes de LMC tratados previamente. La mediana de la duración del tratamiento fue de 14,1 meses (intervalo: de 0,3 a 24,7 meses), de 61,6 meses (intervalo: de 0,03 a 99,6 meses), de 11,1 meses (intervalo: de 0,03 a 123,3 meses), de 30,2 meses (intervalo: de 0,3 a 85,6 meses) y de 5,7 meses (intervalo: de 0,07 a 17,8 meses), respectivamente. Los análisis de seguridad incluyeron datos de un estudio de extensión en curso. Se notificó al menos una reacción adversa de grado variable de toxicidad en 1.240 (97,5%) pacientes. Las reacciones adversas notificadas más frecuentemente en ³ 20% de los pacientes fueron diarrea (78,1%), náuseas (40,8%), trombocitopenia (34,9%), dolor abdominal (34,0%), vómitos (33,0%), erupción cutánea (31,5%), anemia (25,6%), pirexia (21,8%), fatiga (21,4%) y elevación de la concentración de ALT (25,0%). Se notificó al menos una reacción adversa de grado 3 o grado 4 en 814 (63,9%) pacientes. Las reacciones adversas de grado 3 o grado 4 notificadas en ³ 5% de los pacientes fueron trombocitopenia (20,3%), anemia (10,2%), neutropenia (10,5%), elevación de la concentración de ALT (12,7%), diarrea (9,6%), erupción (5,0%), elevación de la concentración de lipasas (8,2%) y elevación de la concentración de AST (5,8%). Tabla de reacciones adversas: Las siguientes reacciones adversas se notificaron en pacientes de los estudios clínicos con bosutinib (Tabla 2). Representan una evaluación de los datos de las reacciones adversas procedentes de 1272 pacientes con LMC en fase crónica recién diagnosticada, con LMC en fase crónica, acelerada o blástica resistentes o intolerantes a tratamientos anteriores o con LLA Ph+ que habían recibido al menos una dosis de bosutinib como agente único. Estas reacciones adversas se presentan conforme al sistema de clasificación por órganos y frecuencia. Las categorías de frecuencia se definen mediante la siguiente convención: muy frecuentes (≥1/10), frecuentes (≥1/100 a < 1/10), poco frecuentes (³1/1.000 a < 1/100), raras (≥1/10.000 a < 1/1.000), muy raras ( < 1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Dentro de cada intervalo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.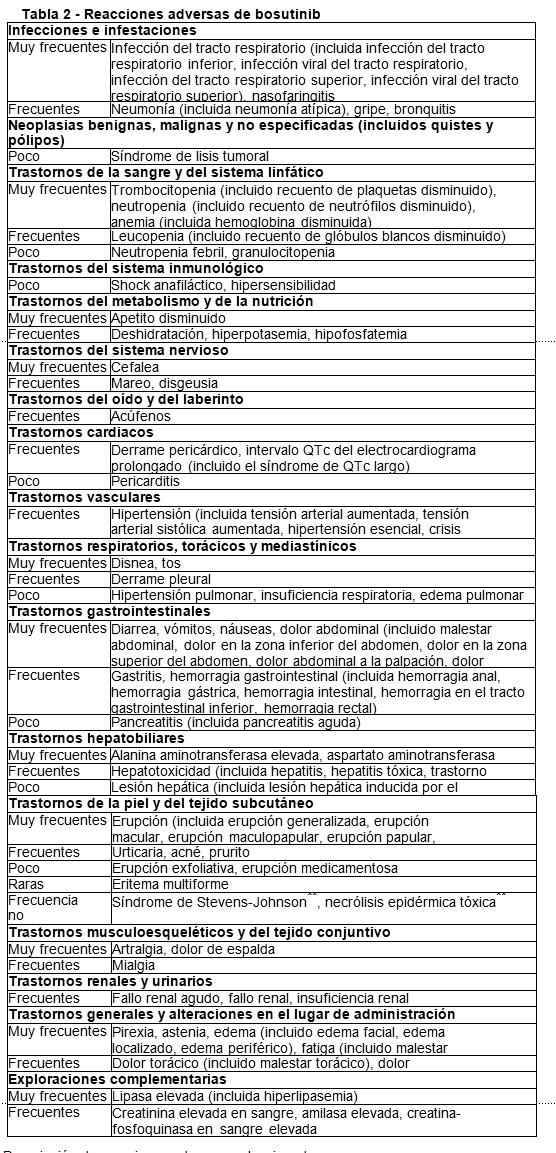
Descripción de reacciones adversas seleccionadas: Las descripciones que se incluyen a continuación están basadas en las observaciones realizadas sobre la seguridad de una población de 1272 pacientes, quienes recibieron al menos una dosis de bosutinib y que o bien padecían LMC en FC recién diagnosticada, o bien LMC en FC, FA o FB resistente o intolerante a tratamientos anteriores, o bien padecían LLA Ph+. Trastornos de la sangre y del sistema linfático: De los 297 (23%) pacientes con notificaciones por reacciones adversas de anemia, a 3 pacientes se les suspendió bosutinib a causa de la anemia. De estos pacientes, 174 (58%) experimentaron toxicidad de grado 1 o 2 como toxicidad máxima, en 96 (32%) pacientes fue de grado 3, y en 27 (9%) pacientes fue de grado 4. En estos pacientes, la mediana del tiempo hasta el primer acontecimiento fue de 28 días (intervalo comprendido entre 1 y 2.633 días) y la mediana de la duración por acontecimiento fue de 15 días (intervalo comprendido entre 1 y 1.529 días). De los 197 (15%) pacientes con notificaciones por reacciones adversas de neutropenia, a 15 pacientes se les suspendió bosutinib a causa de la neutropenia. Sesenta y tres (32%) pacientes experimentaron acontecimientos de grado 1 o 2 como toxicidad máxima. La neutropenia de grado 3 como toxicidad máxima fue experimentada por 90 (46%) pacientes, y fue de grado 4 en 44 (22%) pacientes. La mediana del tiempo hasta el primer acontecimiento fue de 59 días (intervalo comprendido entre 27 y 505 días) y la mediana de la duración por acontecimiento fue de 15 días (intervalo comprendido entre 1 y 913 días). De los 445 (35%) pacientes con notificaciones por reacciones adversas de trombocitopenia, a 41 (9%) pacientes se les suspendió el tratamiento con bosutinib a causa de la trombocitopenia. Ciento ochenta y seis (42%) pacientes experimentaron acontecimientos de Grado 1 o 2 como toxicidad máxima. La trombocitopenia de grado 3 como toxicidad máxima fue experimentada por 161 (36%) pacientes, y fue de grado 4 en 98 (22%) pacientes. En los pacientes con reacciones adversas de trombocitopenia, la mediana del tiempo hasta el primer acontecimiento fue de 28 días (intervalo comprendido entre 1 y 1.688 días) y la mediana de la duración por acontecimiento fue de 15 días (intervalo comprendido entre 1 y 1.762 días). Trastornos hepatobiliares: En los pacientes con notificaciones por reacciones adversas de elevaciones de ALT o de AST (todos los grados), la mediana del tiempo observado hasta la aparición de estos trastornos fue de 29 días, con un intervalo comprendido entre 1 y 2.465 días hasta la aparición de la elevación de la ALT y la AST. La mediana de la duración por acontecimiento fue de 18 días (intervalo comprendido entre 1 y 775 días) y de 15 días (intervalo comprendido entre 1 y 803) para la ALT y la AST, respectivamente. A lo largo de todo el programa de desarrollo se produjo, sin causas alternativas, una elevación simultánea de las concentraciones de transaminasas ≥3 x LSN y de bilirrubina > 2 x LSN con la fosfatasa alcalina < 2 x LSN en 1/1.611 ( < 0,1%) de los sujetos tratados con bosutinib. Esta observación tuvo lugar en un estudio de bosutinib en combinación con letrozol en una paciente con cáncer de mama metastásico. Reactivación del virus de la hepatitis B: Se ha notificado reactivación de la hepatitis B en relación con los TKIs BCR-ABL. En algunos casos se ha producido insuficiencia hepática aguda o hepatitis fulminante que ha dado lugar a trasplante de hígado o a un desenlace mortal. Trastornos gastrointestinales: De los 994 (78%) pacientes que experimentaron diarrea, a 10 se les retiró el tratamiento con bosutinib a causa de ese acontecimiento. Se administraron medicamentos concomitantes para tratar la diarrea en 662 (66%) pacientes. La diarrea fue de grado 1 o 2 como toxicidad máxima en el 88% de los pacientes y de grado 3 en el 12% de los pacientes; 1 paciente ( < 1%) sufrió un acontecimiento de grado 4. En los pacientes con diarrea, la mediana del tiempo hasta el primer acontecimiento fue de 2 días (intervalo comprendido entre 1 y 2.415 días) y la mediana de la duración de la diarrea de cualquier grado fue de 2 días (intervalo comprendido entre 1 y 2.511 días). De los 994 pacientes con diarrea, a 180 (18%) pacientes se les interrumpió el tratamiento, y a 170 (94%) de ellos se les volvió a instaurar el tratamiento con bosutinib. De los pacientes a quienes se les reinstauró el tratamiento, 167 (98%) no presentaron más acontecimientos diarreicos o no se les suspendió bosutinib a causa de acontecimientos diarreicos posteriores. Trastornos cardiacos: Cuatro pacientes (0,3%) sufrieron una prolongación del intervalo QTcF (superior a 500 milisegundos). Nueve (0,8%) pacientes experimentaron un aumento del QTcF desde el nivel basal que excedió los 60 milisegundos. En los estudios clínicos no se incluyeron pacientes con enfermedades cardiovasculares no controladas o importantes, incluyendo prolongación del QTc en los electrocardiogramas basales. Notificación de sospechas de reacciones adversas: Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas.
Advertencias.
Alteraciones de la función hepática: El tratamiento con bosutinib se asocia con aumentos en las transaminasas séricas (alanina aminotransferasa [ALT], aspartato aminotransferasa [AST]). Dichos aumentos se produjeron generalmente al comienzo del tratamiento (de los pacientes que experimentaron un aumento de transaminasas de cualquier grado, > 80% de dichos pacientes experimentaron el primer evento dentro de los 3 primeros meses). Los pacientes en tratamiento con bosutinib deben hacerse pruebas de la función hepática antes del inicio del tratamiento y mensualmente durante los 3 primeros meses de tratamiento, y según indicación clínica. Los pacientes con aumento de las transaminasas han de manejarse mediante interrupción temporal del tratamiento con bosutinib (considerando que tras recuperación a Grado 1 o al nivel basal puede hacerse una reducción de la dosis), o mediante suspensión definitiva de bosutinib. Los aumentos de transaminasas, especialmente si tienen lugar con incrementos concomitantes de bilirrubina, pueden constituir una señal temprana de lesión hepática inducida por el medicamento, por lo que estos pacientes han de ser manejados adecuadamente Diarrea y vómitos: El tratamiento con bosutinib se asocia con diarrea y vómitos, por lo que pacientes con alteraciones gastrointestinales clínicamente significativas recientes o en curso deberían utilizar este medicamento con precaución y únicamente tras una cuidadosa evaluación del riesgo-beneficio, ya que pacientes de estas características fueron excluidos de los ensayos clínicos. A los pacientes con diarrea y vómitos se les debe proporcionar el tratamiento habitual, que incluye un medicamento antidiarreico o antiemético y/o la reposición de líquidos. Además, la diarrea y los vómitos pueden controlarse interrumpiendo temporalmente el tratamiento con bosutinib, reduciendo la dosis o con la retirada definitiva de bosutinib. El agente antiemético, domperidona, tiene el potencial de prolongar el intervalo QT (QTc) y de inducir arritmias - "torsade de pointes"; por ello, debe evitarse coadministrar domperidona. Sólo debe usarse en caso de que el resto de medicamentos no resulten eficaces. En situaciones de este tipo es obligatorio realizar una evaluación individualizada de la relación riesgo/beneficio, y debe vigilarse la posible aparición de una prolongación del intervalo QTc en los pacientes. Mielosupresión: El tratamiento con bosutinib se asocia con mielosupresión, definida como anemia, neutropenia y trombocitopenia. Deben realizarse recuentos sanguíneos completos semanalmente durante el primer mes, y a partir de ahí, mensualmente, o según indicación clínica. La mielosupresión debe o puede controlarse interrumpiendo temporalmente el tratamiento con bosutinib, reduciendo la dosis o con la retirada definitiva de bosutinib. Retención de líquidos: El tratamiento con bosutinib puede asociarse a una retención de líquidos, incluyendo derrame pericárdico, derrame pleural, edema pulmonar y/o edema periférico. Los pacientes se deben monitorizar y controlar utilizando el tratamiento habitual. Además, la retención de líquidos puede controlarse interrumpiendo temporalmente el tratamiento con bosutinib, reduciendo la dosis o con la retirada definitiva de bosutinib. Lipasa sérica: Se ha observado aumento de la lipasa sérica. Se recomienda proceder con precaución en los pacientes con antecedentes de pancreatitis. En caso de que los aumentos de la lipasa se presenten acompañados de síntomas abdominales, debe interrumpirse el tratamiento con bosutinib y adoptar las medidas diagnósticas que se consideren adecuadas para excluir pancreatitis. Infecciones: Bosutinib puede predisponer a los pacientes a infecciones bacterianas, fúngicas, víricas o protozoarias. Potencial proarrítmico: Se ha observado prolongación de la repolarización cardiaca ventricular (intervalo QTc) según la lectura automática realizada por el electrocardiógrafo, sin arritmia concomitante. Bosutinib debe administrarse con precaución a los pacientes con antecedentes o predisposición a la prolongación del intervalo QTc, que tengan una cardiopatía no controlada o significativa incluyendo infarto de miocardio reciente, insuficiencia cardiaca congestiva, angina inestable o bradicardia clínicamente significativa, o que estén recibiendo medicamentos con un efecto conocido de prolongación del QTc (por ejemplo, antiarrítmicos u otros productos que puedan prolongar el QTc). La presencia de hipopotasemia e hipomagnesemia puede potenciar este efecto. Resulta aconsejable realizar una monitorización para ver si aparece algún efecto en el QTc, y se recomienda realizar un electrocardiograma (ECG) basal antes de iniciar el tratamiento con bosutinib, y cuando esté clínicamente indicado. Antes de administrar bosutinib se debe corregir la hipopotasemia o la hipomagnesemia, y se han de monitorizar periódicamente durante el tratamiento. Insuficiencia renal: El tratamiento con bosutinib puede provocar una disminución clínicamente significativa de la función renal en pacientes con LMC. Se ha observado una disminución a lo largo del tiempo de la tasa de filtración glomerular estimada (TFGe) en pacientes que recibieron tratamiento con bosutinib en ensayos clínicos. En pacientes con LMC en FC recién diagnosticada tratados con 400 mg, la mediana de disminución de la TFGe en relación con el valor basal fue de 4,9 ml/min/1,73 m2 a los 3 meses, 9,2 ml/min/1,73 m2 a los 6 meses y 11,1 ml/min/1,73 m2 a los 12 meses. Los pacientes no tratados previamente con LMC tratados con 500 mg mostraron una mediana de disminución de TFGe de 5,1 ml/min/1,73 m2 a los 3 meses, de 9,2 ml/min/1,73 m2 a los 12 meses y de hasta 16,3 ml/min/1,73 m2 hasta los 5 años de seguimiento para pacientes en tratamiento. Los pacientes con LMC tratada previamente y en fase avanzada tratados con 500 mg mostraron una mediana de disminución de TFGe de 5,3 ml/min/1,73 m2 a los 3 meses, de 7,6 ml/min/1,73 m2 a los 12 meses y de hasta 10,9 ml/min/1,73 m2 en un periodo de hasta 4 años de tratamiento. Es importante evaluar la función renal antes de iniciar el tratamiento y supervisarla minuciosamente durante el tratamiento con bosutinib, prestando especial atención en el caso de aquellos pacientes que tienen una insuficiencia renal preexistente o factores de riesgo de disfunción renal, incluyendo el uso concomitante de medicamentos que puedan provocar nefrotoxicidad, como diuréticos, inhibidores de la enzima convertidora de angiotensina (ECA), bloqueadores de los receptores de la angiotensina y medicamentos antiinflamatorios no esteroideos (AINEs). En un estudio sobre la insuficiencia renal, las exposiciones a bosutinib se aumentaron en pacientes con la función renal moderada y gravemente afectada. Se recomienda reducir la dosis en pacientes con insuficiencia renal moderada o grave. En los estudios de LMC se excluyó a los pacientes con una creatinina sérica > 1,5 veces el LSN. De acuerdo a un análisis de las características farmacocinéticas, se observó que, al inicio del tratamiento, durante los estudios se produjo un aumento de la exposición (ABC) en los pacientes con una insuficiencia renal moderada y grave. Los datos clínicos son muy limitados (n = 3) para pacientes con LMC e insuficiencia renal moderada que reciben una dosis aumentada de 600 mg de bosutinib. Raza asiática: Según los análisis farmacocinéticos poblacionales, la población asiática tuvo un aclaramiento menor, lo que aumentó la exposición. Por lo tanto, estos pacientes se deben someter a una estrecha monitorización para detectar reacciones adversas, especialmente en caso de aumento de la dosis. Reacciones cutáneas graves: Bosutinib puede provocar reacciones cutáneas graves, tales como el síndrome de Stevens-Johnson y la necrólisis epidérmica tóxica. El tratamiento con bosutinib se debe interrumpir de forma permanente en los pacientes que experimenten una reacción cutánea grave durante el tratamiento. Síndrome de lisis tumoral: Debido a la posible aparición del síndrome de lisis tumoral (SLT), se recomienda la corrección de la deshidratación clínicamente significativa y el tratamiento de los niveles altos de ácido úrico antes de iniciar el tratamiento con bosutinib. Reactivación del virus de la hepatitis B: Se ha producido reactivación de la hepatitis B en pacientes que son portadores crónicos de este virus después de que los pacientes hayan recibido TKIs BCR-ABL. En algunos casos se produjo insuficiencia hepática aguda o hepatitis fulminante que dio lugar a un trasplante de hígado o a un desenlace mortal. Los pacientes se deben someter a pruebas para detectar la infección por VHB antes de comenzar el tratamiento con bosutinib. Se debe consultar a expertos en enfermedades hepáticas y en el tratamiento de la hepatitis B antes de comenzar el tratamiento en pacientes con una serología positiva para hepatitis B (incluyendo a los pacientes con enfermedad activa) y pacientes que den un resultado positivo en una prueba de infección por VHB durante el tratamiento. Los portadores del VHB que necesiten tratamiento con bosutinib se deben someter a una estrecha monitorización para detectar signos y síntomas de infección activa por VHB a lo largo de todo el tratamiento y durante varios meses después de finalizar el tratamiento. Inhibidores del citocromo P-450 (CYP)3A: Debe evitarse el uso concomitante de bosutinib con inhibidores potentes o moderados del CYP3A, ya que se produciría un aumento de la concentración plasmática de bosutinib. Se recomienda seleccionar, si es posible, un medicamento concomitante alternativo cuyo potencial de inhibición del CYP3A sea nulo o mínimo. Si durante el tratamiento con bosutinib resulta necesario administrar un inhibidor potente o moderado del CYP3A, hay que plantearse interrumpir el tratamiento con bosutinib o reducir la dosis de bosutinib. Inductores del CYP3A: Debe evitarse el uso concomitante de bosutinib con inductores potentes o moderados del CYP3A, ya que se produciría una disminución de la concentración plasmática de bosutinib. Efecto de los alimentos: Debe evitarse el consumo de productos que contengan pomelo, incluyendo el zumo de pomelo, así como de otros alimentos con un efecto conocido de inhibición del CYP3A. Contenido en sodio: Este medicamento contiene menos de 1 mmol de sodio (23 mg) por comprimido de 100 mg, 400 mg o 500 mg. Se puede informar a los pacientes con dietas bajas en sodio que este medicamento está esencialmente "exento de sodio".
Presentación.
Bosulif Comprimidos Recubiertos 100 mg. Bosulif Comprimidos Recubiertos 400 mg. Bosulif Comprimidos Recubiertos 500 mg.