BRITAXOL®
BMS
Antineoplásico.
Descripción.
BRITAXOL® (paclitaxel) solución inyectable para infusión intravenosa es una solución viscosa transparente, incolora a levemente amarilla. Es proporcionada como una solución no acuosa para dilución con un líquido parenteral adecuado antes de proceder a la infusión intravenosa. BRITAXOL® está disponible en viales de dosis múltiples de 30 mg (5 mL), 100 mg (16,7 mL) y 300 mg (50 mL). Cada ml de la solución estéril no pirógena contiene 6 mg de paclitaxel, Cremophor EL* purificado (aceite de ricino polioxietilado) y alcohol deshidratado, Ph. Eur. Paclitaxel es un producto de origen natural con actividad antitumoral. BRITAXOL® contiene paclitaxel el cual se obtiene por un proceso de biosíntesis natural de fermentación. El nombre químico para paclitaxel es 5b,20-Epoxi-1,2a,4,7b,10b, 13a-hexahidroxitax-11-eno-9-ona 4,10-diacetato 2-benzoato 13-éster con (2R,3S)-N-benzoil-3-fenilisoserina.
Paclitaxel tiene la siguiente fórmula estructural: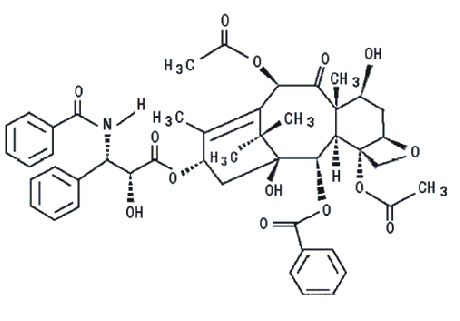
Paclitaxel es un polvo cristalino blanco a blanco cremoso, con la fórmula empírica de C47H51NO14 y un peso molecular de 853.9. Es altamente lipofílico, insoluble en agua y funde a alrededor de 216-217°C.
Farmacología.
Farmacología clínica: Paclitaxel es un agente antimicrotubular novedoso que fomenta la reunión de microtúbulos a partir de dímeros tubulínicos y estabiliza los microtúbulos al evitar la despolimerización. Esta estabilidad da lugar a la inhibición de la reorganización dinámica normal de la red de microtúbulos lo cual es esencial para la interfase vital y las funciones celulares mitóticas. Además, paclitaxel induce la formación de conjuntos anormales o "haces" de microtúbulos a través del ciclo celular y ásteres múltiples de microtúbulos durante la mitosis. Después de la administración intravenosa de BRITAXOL®, las concentraciones plasmáticas de paclitaxel disminuyeron de manera bifásica. La disminución inicial rápida representa la distribución al compartimiento periférico y la eliminación del fármaco. La última fase se debe, en parte, al eflujo relativamente lento de paclitaxel desde el compartimiento periférico. Los parámetros farmacocinéticos de paclitaxel después de infusiones de 3 y 24 horas de BRITAXOL® a niveles posológicos de 135 y 175 mg/m2 fueron determinados en un estudio al azar de Fase 3 en pacientes con cáncer del ovario, y están resumidos en el siguiente cuadro: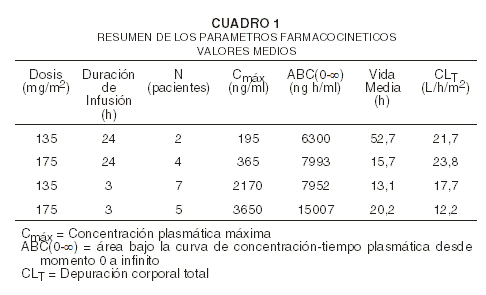
Pareció ser que con la infusión de 24 horas de BRITAXOL®, un incremento del 30% en la posología (135 mg/m2 versus 175mg/m2) aumentó la Cmáx en un 87%; mientras que el ABC(0-inf) continuó proporcional. Sin embargo, con una infusión de 3 horas, para un incremento del 30% de la posología, la Cmáx y el ABC(0-inf) fueron incrementados en un 68% y 89%, respectivamente. El volumen medio aparente de distribución en una condición estable, con la infusión de 24 horas de BRITAXOL®, osciló de 227 a 688 L/m2, indicando una distribución extravascular extensa y/o conjugación a tejidos que tiene paclitaxel. La farmacocinética de paclitaxel también fue evaluada en pacientes adultas con cáncer, quienes recibieron dosis únicas de 15-135 mg/m2 dadas por medio de infusiones de 1 hora (n=15), 30-275 mg/m2 dadas por medio de infusiones de 6 horas (n=36), y 200-275 mg/m2 dadas por infusiones de 24 horas (n=54) en los estudios de fases 1 y 2. Los valores para CLT y el volumen de distribución fueron consistentes con los hallazgos en el estudio de Fase 3. No ha sido estudiada la farmacocinética de BRITAXOL® en pacientes que padecen de sarcoma de Kaposi relacionado al SIDA. Los estudios in vitro sobre la conjugación a proteínas séricas humanas, usando concentraciones de paclitaxel entre 0,1 a 50 mg/ml, indican una conjugación del 89 al 98% del agente; la presencia de cimetidina, ranitidina, dexametasona o difenhidramina no afectó la conjugación proteica de paclitaxel. Después de la administración intravenosa de dosis de 15-275mg/m2 de BRITAXOL® en infusiones de 1, 6 o 24 horas, los valores promedio por recuperación urinaria de fármaco sin cambio variaron de 1,3 a 12,6% de la dosis, indicando un aclaramiento no renal extenso. En cinco pacientes a los que se les administró una dosis de 225 o 250 mg/m2 de BRITAXOL® marcados radiactivamente en una infusión de 3 horas, un promedio de 71% de la radiactividad fue excretada en las heces, en 120 horas, y el 14% fue recobrado en la orina. La recuperación total de radiactividad estuvo en un rango de 56 a 101% de la dosis. Paclitaxel representó un promedio de 5% de la radiactividad administrada recobrada en las heces, mientras que los metabolitos, primariamente 6-hidroxipaclitaxel, contaron para el balance. Estudios in vitro con microsomas del hígado humano y cortes tisulares mostraron que paclitaxel fue metabolizado primariamente a 6-hidroxipaclitaxel por el citocromo P450 isoenzima CYP2C8; y dos metabolitos menores, 3'-p-hidroxipaclitaxel y 6,3'-p-dihidroxipaclitaxel, por CYP3A4. In vitro, el metabolismo de paclitaxel a 6-hidroxipaclitaxel fue inhibido por un número de agentes (ketoconazol, verapamil, diazepam, quinidina, dexametasona, ciclosporina, tenipósido, etopósido, y vincristina), pero las concentraciones usadas excedieron aquellas encontradas in vivo siguiendo dosis terapéuticas normales. Testosterona, 17-etinil estradiol, ácido retinoico, y quercetin, un inhibidor específico de CYP2C8, también inhibió la formación de 6-hidroxipaclitaxel in vitro. La farmacocinética de paclitaxel puede también ser alterada in vivo como un resultado de interacciones con compuestos que son sustratos, inductores, o inhibidores de CYP2C8 y/o CYP3A4 (ver Precauciones: Interacciones con fármacos). La disposición y la toxicidad de una infusión de 3 horas de paclitaxel fueron evaluadas en 35 pacientes con diferentes grados de alteración de la función hepática. En relación con los pacientes con bilirrubina normal, la exposición plasmática de paclitaxel en pacientes con un valor de bilirrubina sérica anormal ≤2 veces el límite superior de lo normal (ULN) y que recibieron 175 mg/m2 estaba aumentada, pero sin un aumento aparente en la frecuencia o la severidad de la toxicidad. En 5 pacientes con bilirrubina sérica total ≥2 veces el ULN, hubo una incidencia mayor, no estadísticamente significativa, de mielosupresión severa aún a una dosis reducida (110 mg/m2), pero sin observar aumento en la exposición plasmática (ver Precauciones: hepática y Dosificación). El efecto de la alteración de la función renal sobre la disposición del paclitaxel no ha sido investigado. No se han investigado formalmente las interacciones posibles de paclitaxel con medicamentos administrados concomitantemente.
Indicaciones.
BRITAXOL® está indicado como terapia de primera línea y subsecuente para el tratamiento de carcinoma avanzado del ovario. Como terapia de primera línea, BRITAXOL® está indicado en combinación con cisplatino. BRITAXOL® está indicado para el tratamiento adyuvante del cáncer nódulo-positivo de mama, el cual es administrado secuencialmente a la quimioterapia estándar de combinación que contiene doxorrubicina. En la prueba clínica, hubo un efecto global favorable sobre las sobrevivencias libre de enfermedad y global en la población total de pacientes con tumores receptores-negativos y receptores-positivos, pero el beneficio ha sido demostrado específicamente con datos disponibles (seguimiento medio de 30 meses) únicamente en pacientes con tumores receptores-negativos de estrógenos y progesterona. BRITAXOL® está indicado para el tratamiento de cáncer de mama después del fracaso de una quimioterapia combinada para la enfermedad metastásica, o recaída después de 6 meses de quimioterapia auxiliar. La terapia previa debiera haber incluido una antraciclina, a menos que esté contraindicada clínicamente. BRITAXOL® en combinación con cisplatino está indicado para el tratamiento de primera línea de cáncer de células no-pequeñas del pulmón en pacientes que no son candidatos para cirugía potencialmente curativa y/o terapia con radiación. BRITAXOL® está indicado como tratamiento de segunda línea del sarcoma de Kaposi relacionado al SIDA.
Dosificación.
Nota: no se recomienda el contacto del concentrado sin diluir con los equipos o dispositivos de PVC plastificados, usados para preparar soluciones para infusión. A fin de reducir al mínimo la exposición de la paciente al plastificador DEHP [di-(2-etilhexil) ftalato], que puede lixiviar de las bolsas o dispositivos de infusión de PVC, las soluciones diluidas de BRITAXOL® deben almacenarse en frascos (de vidrio o polipropileno) o bolsas de material plástico (polipropileno, poliolefina) y administrarse a través de juegos de administración forrados de polietileno. Todas las pacientes deben recibir medicamentos previos a la administración de BRITAXOL® para evitar las reacciones graves de hipersensibilidad. Tales medicamentos previos pueden comprender dexametasona, a 20 mg por vía oral, administrada aproximadamente 12 y 6 horas antes del BRITAXOL®; difenhidramina (o su equivalente) a 50 mg por IV, 30 a 60 minutos antes del BRITAXOL®; y cimetidina (300 mg) o ranitidina (50 mg) por IV, 30 a 60 minutos antes del BRITAXOL®. Para las pacientes que padecen de carcinoma del ovario, se recomiendan los siguientes regímenes: 1. Para pacientes sin tratamiento previo con carcinoma del ovario, se puede dar cada tres semanas uno de los siguientes regímenes recomendados. Al seleccionar el régimen apropiado, se deben considerar las diferencias en toxicidad (ver cuadro Reacciones adversas: Experiencias sobre eventos adversos específicos de enfermedad). BRITAXOL® administrado por vía intravenosa por 3 horas a una dosis de 175 mg/m2 seguido por cisplatino a una dosis de 75 mg/m2; o BRITAXOL® administrado por vía intravenosa por 24 horas a una dosis de 135 mg/m2 seguido por cisplatino a una dosis de 75 mg/m2. En pacientes previamente tratadas con quimioterapia para el carcinoma del ovario, BRITAXOL® ha sido usado en varias dosis y programas de dosificación; sin embargo, el régimen óptimo todavía no está claro. El régimen recomendado es de 135 mg/m2 o 175 mg/m2 de BRITAXOL® administrado por vía intravenosa durante 3 horas cada tres semanas. Para pacientes que padecen de carcinoma de mama, se recomiendan los siguientes regímenes: para el tratamiento adyuvante del cáncer nódulo-positivo de mama, el régimen recomendado es BRITAXOL® a una dosis intravenosa de 175 mg/m2, durante 3 horas, cada 3 semanas por cuatro cursos, administrados secuencialmente a la quimioterapia de combinación que contiene doxorrubicina. La prueba clínica utlizó cuatro cursos de doxorrubicina y ciclofosfamida (ver Estudios clínicos: carcinoma de mama). Después del fracaso de la quimioterapia inicial para la enfermedad metastásica o la recaída dentro de los siguientes 6 meses de la quimioterapia adyuvante, ha demostrado ser efectivo el BRITAXOL® a una dosis de 175 mg/m2, administrado por vía intravenosa durante 3 horas cada 3 semanas, tal y como se ha demostrado que es efectivo. Para pacientes con carcinoma de células no-pequeñas del pulmón, el régimen recomendado es BRITAXOL® administrado por vía intravenosa durante 24 horas, cada 3 semanas, una dosis de 135 mg/m2 seguida de 75 mg/m2 de cisplatino. Para pacientes con sarcoma de Kaposi relacionado al SIDA, se recomienda BRITAXOL® administrado a una dosis de 135 mg/m2, dado por vía intravenosa durante 3 horas, cada 3 semanas, o a una dosis de 100 mg/m2 dada por vía intravenosa durante 3 horas, cada 2 semanas (la intensidad de la dosis es de 45-50 mg/m2/semana). En las dos pruebas clínicas que evaluaron estos programas y horarios de tratamiento, el primer programa (135 mg/m2 cada 3 semanas) fue más tóxico que el segundo. Además, todos los pacientes con baja condición de desempeño fueron tratados con el segundo programa (100 mg/m2 cada 2 semanas). Basado en la inmunosupresión en los pacientes con enfermedad avanzada de VIH, se recomiendan las siguientes modificaciones en estos pacientes: reducir la dosis de dexametasona, como uno de los tres fármacos de premedicación, a 10 mg PO (en vez de 20 mg PO); iniciar o repetir el tratamiento con BRITAXOL® sólo si el conteo de neutrófilos es de por lo menos 1000 células/mm3; reducir la dosis de los cursos subsecuentes de BRITAXOL® en un 20% para pacientes que experimentan neutropenia severa (neutrófilos < 500 células/mm3 durante una semana o más); e iniciar un factor hematopoyético de crecimiento (G-CSF) concomitante, tal y como está indicado clínicamente. Para la terapia de pacientes con tumores sólidos (del ovario, de mama y de NSCLC), no deberán repetirse los cursos de BRITAXOL® hasta que el conteo de neutrófilos esté en por lo menos 1500 células/mm3 y el conteo de plaquetas esté en por lo menos 100.000 células/mm3. No deberá darse BRITAXOL® a pacientes que tienen sarcoma de Kaposi relacionado al SIDA si el conteo inicial o subsecuente de neutrófilos es menor a 1000 células/mm3. A los pacientes que experimentan neutropenia severa (neutrófilos < 500 células/mm3 durante una semana o más) o neuropatía periférica severa durante la terapia con BRITAXOL®, se les deberá reducir la dosis en un 20% para los cursos subsecuentes de BRITAXOL®. La incidencia de neurotoxicidad y la severidad de la neutropenia se incrementan con la dosis. Alteración de la función hepática: los pacientes con alteración de la función hepática pueden tener un mayor riesgo de toxicidad, particularmente mielosupresión grado III-IV (ver Farmacología clínica y Precauciones: hepática). Las recomendaciones para el ajuste de dosis para el primer curso de terapia son mostradas en el cuadro 17, tanto para la infusión de 3 horas como para la de 24 horas. Reducciones adicionales de la dosis en cursos subsecuentes debieran estar basadas en la tolerancia individual. Los pacientes deben ser controlados muy de cerca por el desarrollo de una mielosupresión marcada.
Precauciones en preparación y administración: BRITAXOL® es un fármaco anticáncer citotóxico y, como con otros compuestos potencialmente tóxicos, debe tenerse precaución al manejar BRITAXOL®. El uso de guantes es recomendado. Si la solución de BRITAXOL® se pone en contacto con la piel, ésta debe lavarse inmediatamente con jabón y agua. Después de la exposición tópica, los eventos han incluido picazón, sensación de quemadura y enrojecimiento. Si BRITAXOL® se pone en contacto con las membranas mucosas, éstas deben ser lavadas con agua. Después de la inhalación, los siguientes eventos han sido reportados: disnea, dolor de pecho, enrojecimiento de ojos, dolor de garganta y náuseas. Dada la posibilidad de extravasación, es aconsejable monitorear de cerca el sitio de infusión, por posible infiltración durante la administración del fármaco (ver Precauciones: reacción en el sitio de inyección). Preparación para la administración intravenosa (paclitaxel): la solución inyectable para infusión intravenosa debe diluirse antes de la infusión. BRITAXOL® debe diluirse en solución de cloruro de sodio al 0,9%, USP; solución de dextrosa al 5%, USP; solución de dextrosa al 5% y solución de cloruro de sodio al 0.9%, USP; o solución de dextrosa al 5% en solución de Ringer hasta una concentración final de 0,3 a 1,2 mg/mL. Las soluciones mantienen la estabilidad física y química durante 27 horas bajo condiciones de temperatura ambiente (aproximadamente 25°C) y a luz ambiente. Los productos farmacológicos parenterales deben examinarse visualmente por presencia de partículas y decoloración antes de su administración, siempre y cuando la solución y el recipiente lo permitan. Durante la preparación, las soluciones pueden mostrar cierto enturbiamiento, que se atribuye al vehículo de la fórmula. No han sido notadas pérdidas significativas de la potencia después de la entrega simulada de la solución a través de tubos IV que contienen un filtro interno (0,22 micrones). Los datos obtenidos por presencia del plastificador extraíble DEHP [di-(2-etilhexil) ftalato] muestran que los niveles aumentan con el tiempo y la concentración, cuando las diluciones son preparadas en recipientes de PVC. Consecuentemente, no es recomendado el uso de recipientes y juegos para la administración de PVC plastificados. Las soluciones de BRITAXOL® deben prepararse y almacenarse en recipientes de vidrio, polipropileno o poliolefina. Deben usarse juegos de administración que no contengan PVC, como los forrados con polietileno. BRITAXOL® debe administrarse a través de un filtro interno con una membrana microporosa no mayor de 0,22 micrones. El uso de dispositivos filtrantes que incorporan tubos cortos de entrada y salida recubiertos con PVC no ha dado lugar a lixiviación significativa de DEHP. El aparato dispensador, Chemo Dispensing Pin, o aparatos similares con puntas no deberían ser usados con BRITAXOL®, ya que pueden causar problemas que dan por resultado pérdida de la esterilidad de la solución de BRITAXOL®. Estabilidad: los viales no abiertos de BRITAXOL® (paclitaxel) solución inyectable para infusión intravenosa son estables hasta la fecha indicada si son guardados a no más de 30°C en el envase original. Ni la congelación, ni la refrigeración afectan adversamente la estabilidad del producto. Con la refrigeración los componentes en el vial de BRITAXOL® pueden precipitarse, pero se volverán a disolver cuando alcancen la temperatura ambiente, con poco o sin agitamiento. En estas circunstancias, no ocurre impacto sobre la calidad del producto. Si la solución permanece turbia o si se nota un precipitado insoluble, deberá ser descartado el vial. Las soluciones para infusión preparadas tal y como se recomienda, son estables bajo condiciones de temperatura ambiente (aproximadamente 25°C) y luz ambiental hasta 27 horas.
Contraindicaciones.
BRITAXOL® está contraindicado en pacientes con antecedentes de reacciones de hipersensibilidad a BRITAXOL® u otros fármacos formulados en Cremophor EL (aceite de ricino polioxietilado). No deberá usarse BRITAXOL® en pacientes que tienen tumores sólidos con conteos iniciales de neutrófilos de < 1500 células/mm3 o en pacientes que tienen sarcoma de Kaposi relacionado al SIDA con conteos iniciales de neutrófilos de < 1000 células/mm3.
Reacciones adversas.
Análisis combinado de las experiencias sobre efectos adversos a partir de estudios con agente único: los datos del siguiente cuadro están basados en la experiencia de 812 pacientes (493 con carcinoma ovárico y 319 con carcinoma de mama) que fueron inscriptas en 10 estudios y quienes recibieron BRITAXOL® como agente único. Doscientas setenta y cinco pacientes fueron tratadas en ocho estudios de Fase 2, con dosis de BRITAXOL® que oscilaron entre 135 y 300 mg/m2, administradas durante 24 horas (en cuatro de estos estudios, fue administrado G-CSF como apoyo hematopoyético). Trescientas un pacientes fueron tratadas en el estudio al azar de carcinoma ovárico de Fase 3, el cual comparó dos dosis (135 ó 175 mg/m2) y dos horarios (3 o 24 horas) de BRITAXOL®. Doscientas treinta y seis pacientes con carcinoma de mama recibieron BRITAXOL® (135 o 175 mg/m2) administrado durante 3 horas en un estudio controlado.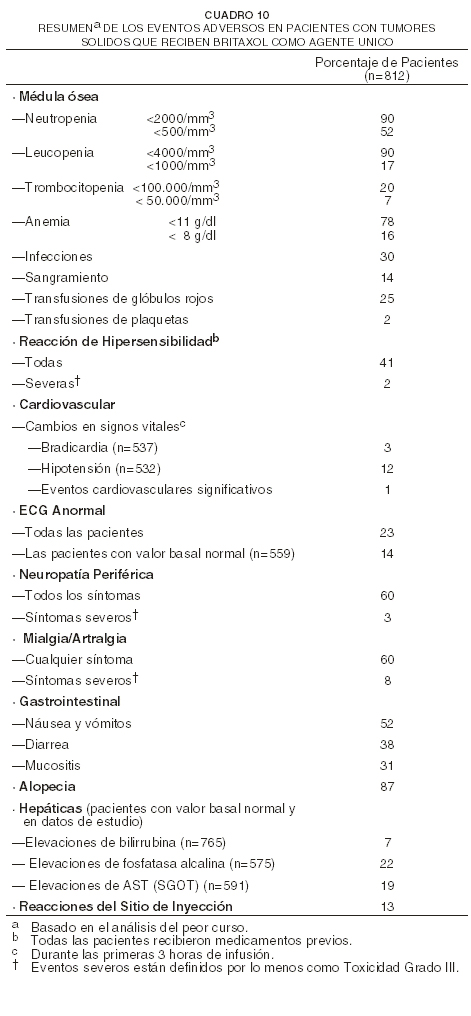
La edad no influyó claramente sobre ninguna de las toxicidades observadas. Experiencias sobre eventos adversos específicos de enfermedad estudio de primera línea en combinación sobre ovarios: para las 1084 pacientes que fueron evaluables en cuanto a seguridad en los estudios de primera línea, fase 3, sobre terapia combinada en ovarios, el cuadro 5 muestra la incidencia de los eventos adversos importantes. Para ambos estudios, el análisis de seguridad se basó en todos los cursos de terapia (seis cursos para el estudio GOG-111 y hasta nueve cursos para el estudio de Intergrupo).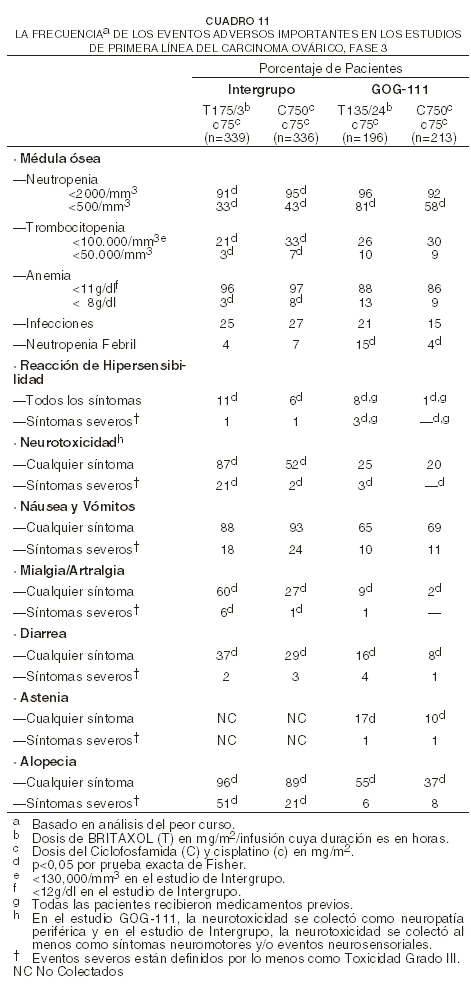
Estudio de segunda línea sobre ovarios: para las 403 pacientes que recibieron BRITAXOL® como agente único durante el estudio de segunda línea Fase 3, sobre carcinoma ovárico, el siguiente cuadro muestra la incidencia de diversos eventos adversos importantes.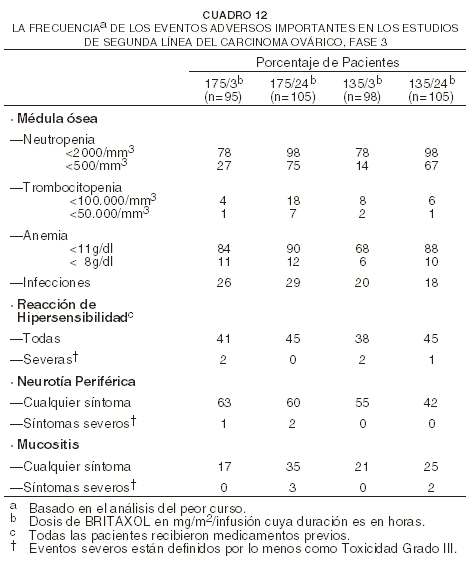
La mielosupresión fue relacionada tanto a posología como a horario, siendo más prominente el efecto del horario. Fue raro el desarrollo de reacciones severas de hipersensibilidad (HSRs); 1% de las pacientes y 0,2% de los cursos en forma global. No fue visto un efecto aparente por posología u horario para las reacciones severas de hipersensibilidad. La neuropatía periférica estuvo relacionada claramente con la posología, pero el horario no pareció afectar la incidencia. Adyuvante de mama: para el estudio Fase 3 de carcinoma adyuvante de mama, el siguiente cuadro muestra la incidencia de eventos adversos severos e importantes para las 3121 pacientes (población total) que fueron evaluables en cuanto a seguridad y para un grupo de 325 pacientes (población inicial) que, según el protocolo del estudio, fueron monitoreadas más intensamente que otras pacientes.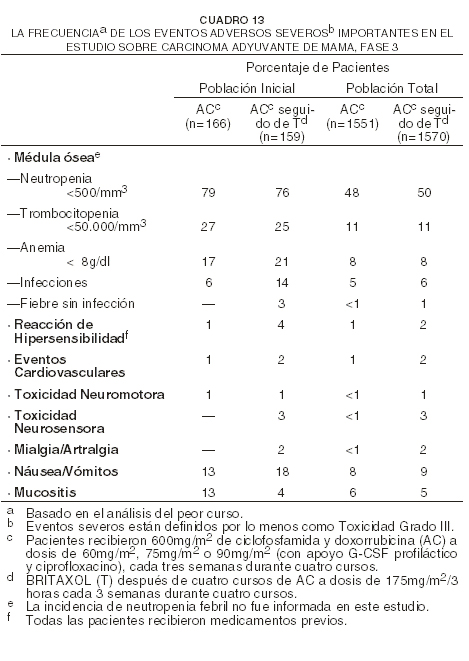
La incidencia de un evento adverso para la población total probablemente representa un subestimado de la incidencia real, ya que los datos fueron recolectados de distinta forma, basándose en el grupo inscripto. Sin embargo, debido a que los datos sobre seguridad fueron recolectados consistentemente a lo largo de los regímenes, la seguridad de la adición secuencial de BRITAXOL® después de una terapia con AC puede compararse con la terapia exclusiva con AC. En comparación a los pacientes que habían recibido únicamente AC, las pacientes que recibieron AC seguido de BRITAXOL® experimentaron más toxicidad neurosensorial Grados III/IV, más mialgia/artralgia Grados III/IV, más dolor neurológico Grados III/IV (5% versus 1%), más síntomas tipo gripe Grados III/IV (5% versus 3%), y más hiperglucemia Grados III/IV (3% versus 1%). Durante los cuatro cursos adicionales de tratamiento con BRITAXOL®, fueron atribuidas dos muertes (0,1%) al tratamiento. Durante el tratamiento con BRITAXOL®, fue informada neutropenia Grados IV para el 15% de las pacientes, toxicidad neurosensorial Grados II/III para el 15%, mialgias Grados II/III para el 23% y alopecia para el 46%. Las incidencias de toxicidades hematológicas severas, infecciones, mucositis y eventos cardiovasculares aumentaron con dosis más altas de doxorrubicina. Cáncer de mama después del fracaso de la quimioterapia inicial: para las 458 pacientes que recibieron BRITAXOL® como agente único en el estudio Fase 3 sobre carcinoma de mama, el siguiente cuadro muestra la incidencia de eventos adversos importantes según el brazo del tratamiento (cada brazo fue administrado con una infusión de 3 horas).
La mielosupresión y la neuropatía periférica estuvieron relacionadas según posología. Hubo una reacción severa de hipersensibilidad observada con una dosis de 135 mg/m2. Estudio de primera línea en combinación sobre NSCLC: en el estudio llevado a cabo por el Grupo Oncológico Cooperativo del Este (ECOG), las pacientes fueron puestas al azar con 135 mg/m2 de BRITAXOL® (T) con infusión de 24 horas en combinación con 75 mg/m2 de cisplatino (c); 250 mg/m2 de BRITAXOL® (T) con infusión de 24 horas en combinación con 75 mg/m2 de cisplatino (c) con apoyo de G-CSF, o 75 mg/m2 de cisplatino (c) el día 1, seguido por 100 mg/m2 de etopósido (VP) los días 1, 2 y 3 (control). El siguiente cuadro muestra la incidencia de eventos adversos importantes.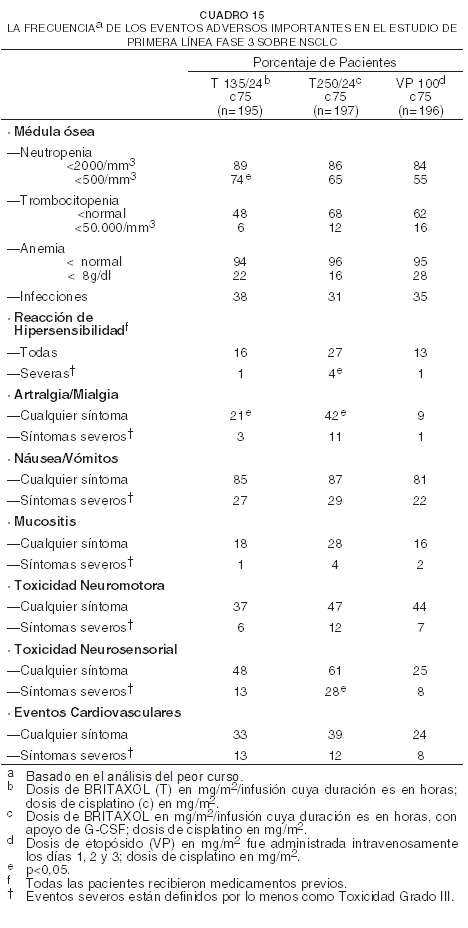
La toxicidad generalmente fue más severa en el brazo de tratamiento con alta dosis de BRITAXOL® (T250/c75) que en el brazo de baja dosis de BRITAXOL® (T135/c75). Comparado con el brazo de cisplatino/etopósido, las pacientes en el brazo de baja dosis de BRITAXOL® experimentaron más artralgia/mialgia de cualquier grado y neutropenia más severa. En este estudio no fue informada la incidencia de neutropenia febril. Sarcoma de Kaposi: el siguiente cuadro muestra la frecuencia de los eventos adversos importantes en los 85 pacientes con sarcoma de Kaposi tratados con dos regímenes distintos de BRITAXOL® como agente único.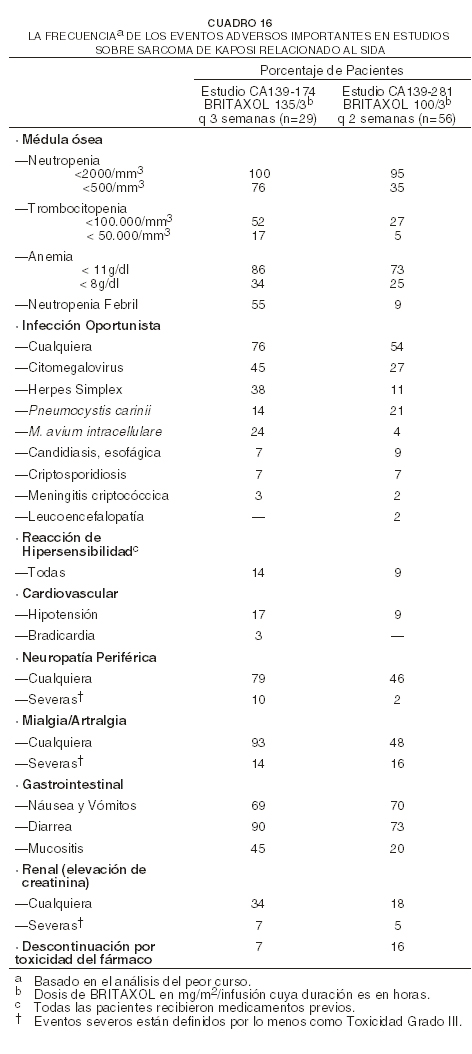
Tal y como fue demostrado en este cuadro, la toxicidad fue más pronunciada en el estudio que utilizó BRITAXOL® a una dosis de 135 mg/m2 cada 3 semanas que en el estudio que utilizó BRITAXOL® a una dosis de 100 mg/m2 cada 2 semanas. Notablemente, fueron más comunes la neutropenia severa (76% versus 35%), la neutropenia febril (55% versus 9%), e infecciones oportunistas (76% versus 54%) con la primera dosis y el primer programa. Deberán tomarse en cuenta las diferencias entre los dos estudios en relación a la escalada de dosis y al uso de factores hematopoyéticos de crecimiento. Nótese también que sólo el 26% de los 85 pacientes en estos estudios recibió tratamiento concomitante con inhibidores de proteasa, cuyo efecto sobre el metabolismo de paclitaxel todavía no ha sido estudiado. Experiencias sobre eventos adversos por sistema corporal: a menos que se indique lo contrario, la siguiente discusión se refiere a la base de datos global sobre seguridad de 812 pacientes que tienen tumores sólidos tratados con BRITAXOL® como agente único, en estudios clínicos. También están descritas las toxicidades que ocurrieron con mayor severidad o frecuencia en las pacientes previamente no tratadas, y que padecían carcinoma ovárico o NSCLC, o quienes recibieron BRITAXOL® en combinación con cisplatino, o en pacientes con cáncer de mama quienes recibieron BRITAXOL® después de doxorrubicina/ciclofosfamida en el cuadro adyuvante, y que ocurrió con una diferencia clínicamente significativa en estas poblaciones. La frecuencia y severidad de los eventos adversos importantes para los estudios Fase 3 sobre carcinoma ovárico, carcinoma de mama, NSCLC, y sobre sarcoma de Kaposi Fase 2, son presentadas arriba en forma tabular y según el brazo del tratamiento. Además, han sido informados eventos raros a partir de experiencias post-mercadeo o de otros estudios clínicos. La frecuencia y severidad de los eventos adversos han sido generalmente similares para pacientes que reciben BRITAXOL® para el tratamiento de carcinoma ovárico, de mama o de pulmón, o sarcoma de Kaposi; pero los pacientes con sarcoma de Kaposi relacionado al SIDA quizás tengan toxicidades hematológicas, infecciones (incluso infecciones oportunistas, ver cuadro 10), y neutropenia febril más frecuentes y más severas. Estos pacientes requieren una intensidad más baja de dosis y cuidado de respaldo. Son descritas las toxicidades cuya ocurrencia fue observada con mayor severidad en la población que padecía de sarcoma de Kaposi y que ocurrieron con una diferencia que fue clínicamente significativa en esta población. Hay una mayor tendencia de incidencia de pruebas de función hepática elevadas y de toxicidad renal en pacientes con sarcoma de Kaposi en comparación a pacientes con tumores sólidos. Hematológicos: la toxicidad principal limitante de la dosis de BRITAXOL® fue la depresión de la médula ósea. La neutropenia, toxicidad hematológica más importante, dependió de la posología y del horario y generalmente fue rápidamente reversible. Entre los pacientes tratados en el estudio de segunda línea Fase 3 sobre ovarios con una infusión de tres horas, los conteos de neutrófilos disminuyeron por debajo de 500 células/mm3 en 14% de las pacientes tratadas con una dosis de 135 mg/m2 en comparación con 27% con una dosis de 175 mg/m2 (p=0,05). En el mismo estudio, fue más frecuente la neutropenia grave ( < 500 células/mm3) con una infusión de 24 horas que con una de 3 horas; la duración de la infusión tuvo mayor impacto sobre la mielosupresión que la posología. La neutropenia no pareció incrementar con la exposición acumulativa y no pareció ser más frecuente ni más severa para las pacientes que habían sido previamente tratadas con terapéutica de radiación. En el estudio en el cual BRITAXOL® fue administrado a pacientes con carcinoma ovárico a una dosis de 135 mg/m2/24 horas, en combinación con cisplatino versus el brazo de control de ciclofosfamida más cisplatino, las incidencias de neutropenia Grado IV y de neutropenia febril fueron significativamente mayores en el brazo de BRITAXOL® más cisplatino que en el brazo control. Ocurrió neutropenia Grado IV en el 81% en el brazo de BRITAXOL® más cisplatino, versus 58% en el brazo de ciclofosfamida más cisplatino, y neutropenia febril ocurrió en el 15% y 4% respectivamente. En el brazo de BRITAXOL®/cisplatino, hubo 35/1074 (3%) eventos con fiebre en los cuales fue informada neutropenia Grado IV en algún momento durante el transcurso. Cuando fue administrado BRITAXOL® seguido de cisplatino a pacientes con NSCLC avanzado en el estudio ECOG, las incidencias de neutropenia Grado IV fueron de 74% (135 mg/m2/24 horas de BRITAXOL® seguido de cisplatino) y 65% (250 mg/m2/24 horas de BRITAXOL® seguido de cisplatino y G-CSF) en comparación con 55% en pacientes que recibieron cisplatino/etopósido. La fiebre fue frecuente (12% de todos los cursos terapéuticos). Ocurrieron episodios infecciosos en 30% de todas las pacientes y en 9% de todos los cursos; estos episodios fueron fatales en 1% de todas las pacientes, e incluyeron sepsis, neumonía y peritonitis. En el estudio ovárico fase 3 de segunda línea, fueron informados episodios infecciosos en 20% y 26% de las pacientes tratadas con una dosis de 135 mg/m2 o 175 mg/m2, dado en infusiones de 3 horas, respectivamente. Las complicaciones infecciosas más frecuentemente informadas fueron las infecciones del tracto urinario y del tracto respiratorio superior. En la población de pacientes con inmunosupresión que tenían la enfermedad de VIH avanzada y sarcoma de Kaposi relacionado al SIDA de bajo riesgo, 61% de los pacientes informó por lo menos una infección oportunista. Se recomienda el uso de terapia de soporte, incluyendo G-CSF, para pacientes que han experimentado una neutropenia severa (ver Dosificación). Se reportó trombocitopenia. El 20% de las pacientes experimentó una caída en su conteo de plaquetas por debajo de 100.000 células/mm3 en por lo menos una vez mientras estuvo en tratamiento; 7% tuvo un conteo de plaquetas < 50.000 células/mm3 al momento de su peor nadir. Fueron informados episodios hemorrágicos en 4% de todos los cursos y en 14% de todas las pacientes, pero la mayor parte de los episodios hemorrágicos fueron localizados y la frecuencia de estos eventos no tuvo relación con las dosis ni el horario de BRITAXOL®. En el estudio de segunda línea, Fase 3 sobre ovarios, fueron informados episodios hemorrágicos en el 10% de las pacientes; ninguna paciente tratada con la infusión de 3 horas recibió transfusiones de plaquetas. En la prueba adyuvante sobre carcinoma de mama, aumentó la incidencia de trombocitopenia severa y transfusiones de plaquetas, con dosis más altas de doxorrubicina. Fue observada anemia (Hb < 11g/dl) en el 78% de todas las pacientes y fue grave (Hb < 8g/dl) en el 16% de los casos. No fue observada relación consistente alguna entre la dosis o el horario y la frecuencia de la anemia. Entre las pacientes con valores basales de hemoglobina normales, el 69% se volvió anémico durante el estudio, pero sólo 7% tuvo anemia grave. Fueron requeridas transfusiones de células sanguíneas en 25% de todas las pacientes y en 12% de aquellas con niveles basales normales de hemoglobina. Reacciones de hipersensibilidad (HSRs): todas las pacientes recibieron medicamentos previos al BRITAXOL® (ver Advertencias y Precauciones: reacciones de hipersensibilidad). La frecuencia y severidad de las reacciones de hipersensibilidad no fueron afectadas por la dosis ni por el horario de la administración de BRITAXOL®. En el estudio de segunda línea Fase 3 de ovarios, la infusión de 3 horas no estuvo asociada con un mayor incremento en las reacciones de hipersensibilidad cuando se comparó con la infusión de 24 horas. Fueron observadas reacciones de hipersensibilidad en 20% de todos los cursos y en 41% de todas las pacientes. Estas reacciones fueron graves en menos del 2% de las pacientes y en 1% de los cursos. No fueron observadas reacciones graves después del curso 3 y los síntomas graves ocurrieron generalmente dentro de la primera hora de infusión con BRITAXOL®. Los síntomas más frecuentes observados durante estas reacciones graves fueron disnea, rubor, dolor en el pecho y taquicardia. También se observó dolor abdominal, dolor en las extremidades, diaforesis e hipertensión. Las reacciones menores de hipersensibilidad consistieron principalmente de rubor (28%), urticaria (12%), hipotensión (4%), disnea (2%), taquicardia (2%) e hipertensión (1%). La frecuencia de las reacciones de hipersensibilidad permaneció relativamente estable durante el período completo de tratamiento. Se han reportado escalofríos, choque, y dolor de espalda en asociación con reacciones de hipersensibilidad. Cardiovascular: la hipotensión ocurrió en 12% de todas las pacientes durante las primeras tres horas de infusión y en 3% de todos los cursos administrados. Durante las primeras tres horas de la infusión ocurrió bradicardia en 3% de todas las pacientes y en 1% de todos los cursos. En el estudio de segunda línea Fase 3 de ovarios, ni la posología ni el horario tuvieron efecto sobre la frecuencia de la hipotensión y bradicardia. Estos cambios en los signos vitales a menudo no causaron síntomas ni se requirió terapia específica o de discontinuación del tratamiento. La frecuencia de hipotensión y bradicardia no fue influenciada por terapia previa con antraciclina. En aproximadamente 1% de todos los pacientes ocurrieron eventos cardiovasculares significativos posiblemente relacionados a BRITAXOL® como agente único. Estos eventos incluyeron síncope, anormalidades rítmicas, hipertensión y trombosis venosa. Uno de los pacientes con síncope, que había sido tratado con 175 mg/m2 de BRITAXOL® durante 24 horas, tuvo hipotensión progresiva y falleció. Las arritmias incluyeron taquicardia ventricular asintomática, bigeminismo y bloqueo AV completo que requirió la colocación de un marcapaso. Entre las pacientes que padecían de NSCLC tratadas con BRITAXOL® en combinación con cisplatino en el estudio de Fase 3, ocurrieron eventos cardiovasculares significativos en 12-13%. Este aumento aparente en los eventos cardiovasculares, posiblemente se debe a un incremento en factores de riesgo cardiovascular en pacientes que padecen de cáncer del pulmón. Fueron comunes las anormalidades del electrocardiograma (ECG) entre pacientes al inicio. Las anormalidades del ECG sobre el estudio usualmente no resultaron en síntomas, no limitaron la posología y no requirieron de intervención. Fueron notadas anormalidades en ECG en 23% de todas las pacientes. Entre las pacientes con un ECG normal previo al ingreso del estudio, 14% de todas las pacientes desarrolló trazos anormales mientras estuvo en el estudio. Las modificaciones más frecuentemente informadas del ECG fueron anormalidades de repolarización no-específicas, bradicardia sinusal, taquicardia sinusal y latidos prematuros. Entre las pacientes con un ECG normal al inicio, la terapia previa con antraciclinas no influyó en la frecuencia de las anormalidades del ECG. Han sido reportados raramente casos de infarto al miocardio. Típicamente se ha reportado, en pacientes quienes han recibido otra quimioterapia, especialmente antraciclinas insuficiencia cardíaca congestiva, incluyendo disfunción cardíaca y reducción de la fracción de eyección ventricular izquierda o fallo ventricular (ver Precauciones: Interacciones con fármacos). Se ha reportado fibrilación auricular y taquicardia supraventricular Respiratorias:Se ha reportado neumonía intersticial, fibrosis pulmonar y embolismo pulmonarSe ha reportado neumonitis por radiación en pacientes recibiendo radioterapia concomitante. Se ha reportado derrame pleural y fallo respiratorio. Neurológica: la evaluación de la toxicidad neurológica se condujo de manera diferente entre los estudios como se evidencia en la información reportada en cada estudio individual (ver cuadros 4-10). Por otra parte, la frecuencia y severidad de las manifestaciones neurológicas se influenciaron por la terapia previa y/o concomitante con agentes neurotóxicos. En general, la frecuencia y severidad de las manifestaciones neurológicas fueron dependientes de la dosis en pacientes que recibían BRITAXOL® como agente único. Se observó neuropatía periférica en 60% de todos los pacientes (3% severa) y en 52% (2% severa) de los pacientes sin neuropatía preexistente. La frecuencia de la neuropatía periférica aumentó con dosis acumulativa. La parestesia comúnmente ocurre en forma de hiperestesia. Los síntomas neurológicos se observaron en 27% de los pacientes luego del primer curso de tratamiento y en 34-51% del curso 2 al 10. La neuropatía periférica fue la causa de la discontinuación de BRITAXOL® en 1% de todos los pacientes. Los síntomas sensoriales mejoran o se resuelven varios meses después de la discontinuación del BRITAXOL®. Las neuropatías preexistentes resultantes de terapias previas no son una contraindicación para BRITAXOL®. En el estudio de Intergrupo de carcinoma ovárico de primera línea (ver cuadro 5), la neurotoxicidad incluyó reportes de eventos neuromotores y neurosensoriales. El régimen con BRITAXOL® 175 mg/m2 dado por una infusión de 3 horas más cisplatino 75 mg/m2 resultó en una incidencia y severidad de neurotoxicidad mayor que el régimen conteniendo ciclofosfamida y cisplatino, 87% (21% severo) versus 52% (2% severo), respectivamente. La duración de la neurotoxicidad de Grado III o IV no se