ECAN-R
SAVAL
Composición.
Cada comprimido recubierto de ECAN-R 80/5/10 contiene: Valsartán 80 mg. Amlodipino (como besilato) 5 mg. Rosuvastatina (como sal cálcica) 10 mg. Excipientes: c.s. Cada comprimido recubierto de ECAN-R 160/5/5 contiene: Valsartán 160 mg. Amlodipino (como besilato) 5 mg. Rosuvastatina (como sal cálcica) 5 mg. Excipientes: c.s. Cada comprimido recubierto de ECAN-R 160/5/10 contiene: Valsartán 160 mg. Amlodipino (como besilato) 5 mg. Rosuvastatina (como sal cálcica) 10 mg. Excipientes: c.s.
Propiedades.
Acción terapéutica: Es un medicamento antihipertensivo gracias al efecto sinergico de Valsartan que bloquea los recetores de angiotensina 2, y de Amplodipino, que bloquea los canales de calcio de las células musculares de los vasos sanguíneos y del corazón. Adicionalmente reduce los niveles de Lipidos en la sangre, como el colesterol y los triglicéridos, gracias a la acción de la Rosusvastatina, la cual inhibe la acción de la enzima HMG-Coa reductasa.
Indicaciones.
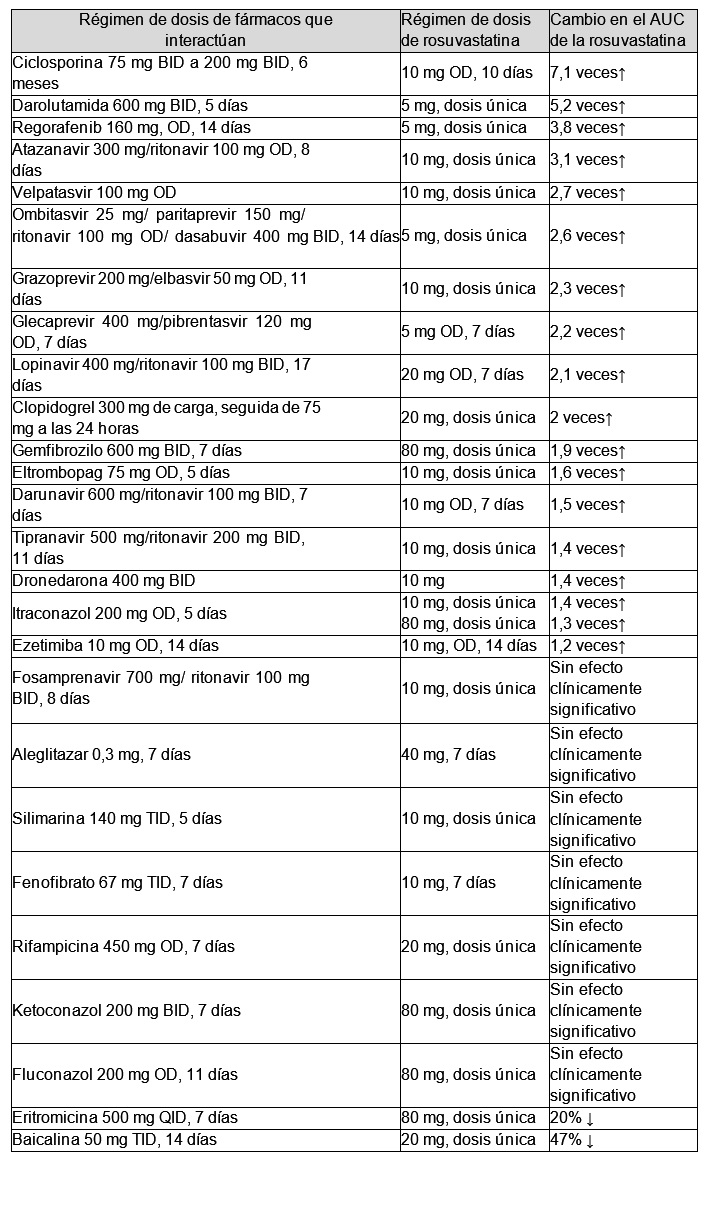
Este medicamento sólo deberá indicarse a quienes necesiten recibir una combinación de dosis fija de amlodipino/valsartán y rosuvastatina al mismo tiempo. La asociación a dosis fijas de VALSARTAN/AMLODIPINO/ROSUVASTATINA está indicada para el tratamiento de pacientes adultos que cursan con hipertensión arterial y dislipidemia. Además, está indicada como reemplazo de terapia en pacientes que están en tratamiento concomitante con los mismos principios activos y en las mismas dosis por separado. Farmacología clínica: Amlodipino: Este antihipertensivo en un antagonista del calcio que inhibe el influjo transmembrana de los iones de calcio hacia el músculo liso vascular y el músculo cardíaco. Este medicamento ofrece un efecto hipotensor al dilatar la arteriola glomerular aferente para aumentar la tasa de filtración glomerular mediante su acción diurética. Valsartán: Este antagonista de la angiotensina II bloquea el efecto vasconstrictor de la angiotensina II, a través de su acción antagonista selectiva y competitiva. Esto genera un efecto hipotensor por la vasodilatación causada por el aumento de la concentración de la renina en sangre. Rosuvastatina: Es un inhibidor de la 3-hidroxi-3 metilglutaril-coenzima A (HMG-CoA) reductasa, la enzima limitante de la velocidad de conversión de HMG-CoA en mevalonato, un precursor del colesterol. Farmacocinética: Se realizó un estudio biofarmacéutico para comparar la biodisponibilidad de este fármaco (amlodipino/valsartán/rosuvastatina) con el uso concomitante de los fármacos (combinación amlodipino/valsartán y rosuvastatina como monofármaco). En total, 53 voluntarios sanos y en ayunas recibieron una sola dosis de una combinación en dosis fija, que contiene 3 principios activos, o medicamentos comercializados, que son amlodipino/valsartán 5/160 mg con rosuvastatina 10 mg (estudio cruzado 2 por 2). Cuando se realizó una evaluación comparativa de los parámetros farmacocinéticos (AUCúltima, Cmáx) mediante el análisis de la concentración plasmática de amlodipino, valsartán, y rosuvastatina, el intervalo de confianza del 90% de la media logarítmica de la diferencia de los parámetros evaluados estaban en el rango de bioequivalencia. (2) Se realizó un estudio farmacocinético comparativo cruzado para evaluar las interacciones farmacológicas entre amlodipino/valsartán y rosuvastatina. En total, 57 voluntarios sanos recibieron la monoterapia de rosuvastatina 20 mg o amlodipino/valsartán 10/160 mg o tratamiento combinado durante 17 días (estudio cruzado 2 por 2). Como resultado de la prueba, no se observaron interacciones farmacocinéticas. Otros estudios farmacocinéticos con otros medicamentos demuestran un aumento de aproximadamente 2 veces en la mediana del AUC y la Cmáx en sujetos asiáticos (japoneses, chinos, filipinos, vietnamitas y coreanos) en comparación con caucásicos. Un análisis farmacocinético poblacional no reveló diferencias clínicamente relevantes en la farmacocinética entre los grupos de población caucásica y negra. Estudios clínicos: Se realizó un estudio aleatorizado, doble ciego, de grupos paralelos, de 8 semanas en 203 pacientes (conjunto de seguridad), que tenían hipertensión y dislipidemia, para una evaluación comparativa de la eficacia y la seguridad de la combinación amlodipino/valsartán/rosuvastatina, o la combinación amlodipino/valsartán, o el tratamiento combinado valsartán/rosuvastatina. Los sujetos recibieron una sola dosis diaria de valsartán 160 mg durante un período de corrección de hábitos de vida de 4 semanas, y los sujetos con una presión arterial sistólica en sedestación no inferior a 140 mg y un nivel de C-LDL adecuado de acuerdo con la clasificación NCEP ATP III al finalizar dicho período fueron asignados aleatoriamente para recibir este fármaco (amlodipino/valsartán/rosuvastatina 10/160/20 mg) o amlodipino/valsartán 10/160 mg o valsartán/rosuvastatina 160/20 mg. Como variables principales, se evaluaron el cambio en la PAS en sedestación (PASsed) con este fármaco en comparación con el grupo valsartán y rosuvastatina, y el cambio (%) en el C-LDL con este fármaco en comparación con el grupo amlodipino/valsartán. Al realizar el análisis de varianza (ANCOVA), el cual ajusta la media basal por las covariables, de las variables principales, que eran el cambio en el valor de la PASsed y del C-LDL desde el inicio, se demostró que el cambio en la PAS en sedestación y el C-LDL en el grupo de prueba fue superior a los grupos comparadores respectivos (p < 0,0001).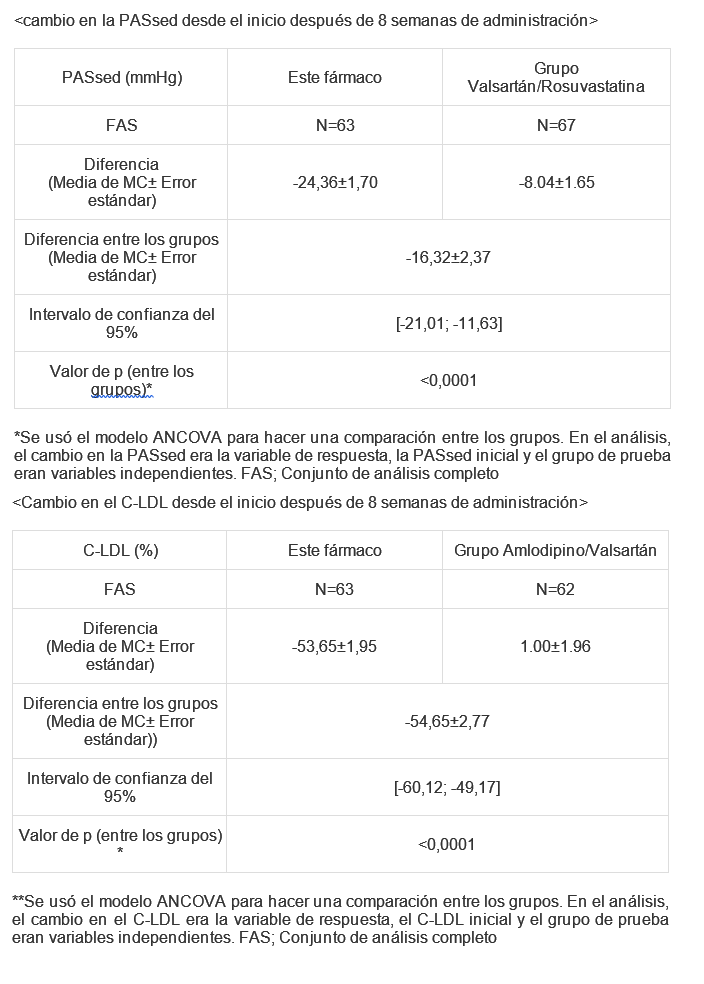
Toxicidad: Toxicidad de dosis repetidas: Se realizó una evaluación toxicológica de dosis repetidas de 13 semanas con el fármaco de prueba (amlodipino/valsartán/rosuvastatina) en ratas, y los resultados no revelaron nuevos cambios toxicológicos además de los cambios toxicológicos ya conocidos de cada sustancia. Toxicidad reproductiva y del desarrollo: No se llevó a cabo un estudio de toxicología reproductiva y del desarrollo para desarrollar la combinación de dosis fijas, pero a continuación se presenta la toxicidad reproductiva y del desarrollo conocida de cada sustancia. Amlodipino: Los estudios reproductivos en ratas y ratones demostraron retraso de la fecha de parto y trabajo de parto prolongado con dosis aproximadamente 50 veces mayores que la dosis máxima recomendada en humanos en mg/kg. No se observó otra toxicidad reproductiva. Valsartán: En ratas, dosis tóxicas para la madre (600 mg/kg/día) durante los últimos días de la gestación y la lactancia dieron lugar a menor sobrevida, menor aumento de peso y retraso en el desarrollo de la cría. El valsartán no tuvo efectos adversos sobre la función reproductiva de ratas macho o hembra con dosis orales de hasta 200 mg/kg/día. Esta dosis es 6 veces la dosis máxima recomendada en humanos en mg/m2. En un estudio de desarrollo embriofetal en ratones, ratas y conejos, se observó fetotoxicidad asociada con toxicidad materna en ratas que recibieron 600 mg/kg/día y conejos que recibieron 10 mg/kg/día. Estas dosis son aproximadamente 0,6 y 18 veces la dosis máxima recomendada en humanos en mg/m2 (los cálculos asumen una dosis oral de 320 mg/día y una paciente de 60 kg). En ratones que recibieron 600 mg/kg/día, no hubo evidencia de toxicidad materna ni fetotoxicidad. Esta dosis era 9 veces la dosis máxima recomendada en humanos en mg/m2. Rosuvastatina: La toxicidad reproductiva fue evidente en ratas, con crías de menor tamaño y peso y menor sobrevida con dosis tóxicas para la madre, con exposiciones sistémicas varias veces por encima del nivel de exposición terapéutico
Dosificación.
La dosis recomendada es un comprimido por día. Se recomienda tomar este medicamento con un poco de agua. De ser posible, se recomienda tomar este medicamente siempre a la misma hora, por ejemplo, antes del desayuno. El uso de este medicamente se limita solo a pacientes adultos, y se deberá considerar una dosis de la combinación de dosis fija de amlodipino/valsartán y rosuvastatina dependiendo del efecto y la tolerancia al medicamento de cada paciente. Para mayor comodidad, los pacientes que reciben la combinación de valsartán/amlodipino y rosuvastatina en comprimidos/cápsulas por separado pueden pasar a recibir este medicamento que contiene las mismas dosis de los componentes. Amlodipino/valsartán en combinación de dosis fija: Se recomienda la titulación individual de la dosis de los componentes (es decir amlodipino o valsartán) antes de cambiar a la combinación de dosis fija. Cuando sea clínicamente apropiado (cuando la presión arterial no esté controlada adecuadamente con monoterapia), podrá considerarse el cambio directo de la monoterapia a la combinación de dosis fija de la siguiente manera:• podrán administrarse 5 mg/80 mg en pacientes cuya presión arterial no esté controlada adecuadamente con amlodipino 5 mg o valsartán 80 mg solos.• podrán administrarse 5 mg/160 mg en pacientes cuya presión arterial no esté controlada adecuadamente con amlodipino 5 mg o valsartán 160 mg solos.• podrán administrarse 10 mg/160 mg en pacientes cuya presión arterial no esté controlada adecuadamente con amlodipino 10 mg o valsartán 160 mg solos o con este medicamento en la concentración 5 mg/160 mg. Insuficiencia renal: No se requiere ajuste de la dosis para pacientes con insuficiencia renal leve a moderada (depuración de creatinina ≥ 10 mL/min). Sin embargo, no deberá usarse para pacientes con insuficiencia renal grave (depuración de creatinina < 10 mL/min) y pacientes en hemodiálisis. Se recomienda controlar los niveles de potasio y la creatinina en pacientes con insuficiencia renal moderada. Insuficiencia hepática: En pacientes con insuficiencia hepática leve a moderada, la dosis diaria máxima recomendada es de 80 mg de valsartán. La combinación de dosis fija de amlodipino/valsartán está contraindicada en pacientes con insuficiencia hepática grave, cirrosis biliar, colestasis, u obstrucción biliar. Ancianos (≥65 años): En pacientes ancianos, se requiere precaución al aumentar la dosis. Rosuvastatina: 1. hipercolesterolemia primaria (tipo IIa que incluye hipercolesterolemia familiar heterocigota), dislipidemia mixta (tipo IIb), disbetalipoproteinemia primaria (hiperlipoproteinemia tipo III) e hipercolesterolemia familiar homocigota. Antes de iniciar el tratamiento con rosuvastatina, se deberá colocar al paciente en una dieta hipocolesterolemiante estándar que deberá continuar durante el tratamiento. La rosuvastatina puede administrarse en cualquier momento del día, con o sin alimentos. La dosis inicial es de 5 mg una vez al día, pero es posible usar la dosis de mantenimiento si es necesaria para reducir más el colesterol LDL. La dosis de mantenimiento es de 10 mg una vez al día, y la mayoría de los pacientes están controlados con esta dosis. La dosis de mantenimiento deberá individualizarse después de 4 o más semanas de acuerdo con el colesterol LDL, el objetivo del tratamiento, y la respuesta del paciente. La dosis diaria máxima es de 20 mg. 2. Ancianos: No es necesario un ajuste de la dosis adicional en relación con la edad. 3. Insuficiencia renal: No es necesario ajustar la dosis para pacientes con insuficiencia renal leve a moderada. La rosuvastatina está contraindicada en pacientes con insuficiencia renal grave. La dosis de 20 mg de rosuvastatina deberá administrarse con cuidado en pacientes con insuficiencia renal moderada. 4. Insuficiencia hepática: No hubo aumento en la exposición sistémica a la rosuvastatina en sujetos con puntuaciones de Child-Pugh ≤7. Sin embargo, se observó aumento de la exposición sistémica en sujetos con puntuaciones de Child-Pugh de 8 y 9. En estos pacientes, deberá considerarse una evaluación de la función renal. No hay experiencia en sujetos con puntuaciones de Child-Pugh superiores a 9. La rosuvastatina está contraindicada en pacientes con enfermedad hepática activa. 5. Raza: Se observó aumento de la exposición sistémica en sujetos asiáticos. La dosis inicial recomendada es de 5 mg para pacientes con ascendencia asiática. La dosis de 40 mg está contraindicada en estos pacientes. 6. Dosis en pacientes con factores de predisposición a miopatía. La dosis inicial recomendada es de 5 mg en pacientes con factores de predisposición a miopatía. La dosis de 40 mg está contraindicada en algunos de estos pacientes. Uso pediatrico: No se han establecido la seguridad y la eficacia en niños menores de 18 años. No se recomienda el uso de este medicamento en niños. Uso en ancianos: No es necesario ajustar la dosis en pacientes ancianos, pero no puede excluirse una mayor sensibilidad en algunos individuos mayores. Por lo tanto, este medicamento debe usarse con cuidado. La depuración de amlodipino en pacientes ancianos mayores de 75 años es reducida, por lo tanto, puede requerirse una dosis inicial más baja y aumentarla lentamente. Dado que, en general, una reducción excesiva de la presión arterial puede causar infartos cerebrales u otros eventos adversos en pacientes ancianos, se la considera inapropiada. Usar este medicamento con cuidado, por ejemplo, empezando con la dosis mínima, etc. En el estudio de farmacodinamia en pacientes ancianos, se observa que la concentración plasmática de valsartán es mayor en ancianos que en más jóvenes. Efectos en las pruebas de laboratorio clínico: En pacientes hipertensos que usaban la combinación valsartán/amlodipino, raramente los resultados de las pruebas de laboratorio presentaron cambios significativos respecto del valor inicial. Se observó un mayor nivel de BUN en el 5,5% de los pacientes que usaban valsartán-amlodipino concomitantemente y en el 5,5% de los pacientes que usaron valsartán como monoterapia en comparación con el 4,5% de los pacientes tratados con placebo. Si bien se informó aumento de los valores de la función hepática con el uso de valsartán, no es necesario un control especial en pacientes con hipertensión esencial (primaria).
Contraindicaciones.
En pacientes con hipersensibilidad a los principios activos o a los derivados de la dihidropiridina. Durante el embarazo y la lactancia en mujeres en edad fértil. En pacientes con insuficiencia hepática grave, cirrosis biliar, obstrucción biliar o colestasis. En pacientes con enfermedad hepática activa, incluidas elevaciones persistentes inexplicables de las transaminasas séricas y cualquier elevación de las transaminasas séricas que exceda 3 veces el límite superior normal (LSN). En pacientes con insuficiencia renal grave (depuración de creatinina < 30 mL/min). Uso concomitante de este medicamento con productos que contengan aliskirén en pacientes con diabetes mellitus o insuficiencia renal moderada-grave (TSG < 60 mL/min/1,73 m2). En pacientes con angioedema hereditario o pacientes que anteriormente tuvieron angioedema con inhibidores de la ECA o antagonistas de la angiotensina II. En pacientes con hiperaldosteronismo primario (no se deberá tratar a los pacientes con hiperaldosteronismo primario con este medicamento ya que su sistema renina-angiotensina no está activado). En pacientes con estenosis aórtica de alto grado. Shock. En pacientes con miopatía. En pacientes que reciben ciclosporina concomitante. La dosis de 40 mg de rosuvastatina está contraindicada en pacientes con factores de predisposición a miopatía/rabdomiólisis. Dichos factores incluyen: - insuficiencia renal moderada (depuración de creatinina < 60 mL/min) - hipotiroidismo - antecedentes personales o familiares de trastornos musculares hereditarios - antecedentes de toxicidad muscular con otro inhibidor de la HMG-CoA reductasa o fibrato - abuso de alcohol - situaciones donde puede ocurrir un aumento en los niveles plasmáticos - pacientes asiáticos - uso concomitante de fibratos.
Embarazo y lactancia.
No se ha establecido la seguridad de este medicamento. Evitar su uso durante el embarazo o la lactancia. Embarazo: Combinación amlodipino/valsartán: No puede excluirse el riesgo de los antagonistas de la angiotensina II (AAII) para el feto. Se informaron casos de morbimortalidad fetal en mujeres embarazadas que usaron inhibidores de la enzima convertidora de angiotensina (fármacos que actúan sobre el sistema renina- angiotensina-aldosterona) cuando cursaban el segundo y tercer trimestre de embarazo. Existe riesgo de teratogenicidad después de la exposición a inhibidores de la ECA durante el primer trimestre de embarazo. Se informaron casos de aborto espontáneo, oligohidramnios, y neonatos con deterioro de la función renal en mujeres embarazadas que usaron accidentalmente valsartán. Al igual que otros medicamentos que actúan sobre el sistema renina-angiotensina-aldosterona, las mujeres que planean quedar embarazadas deberán evitar el uso de la combinación amlodipino/valsartán. Los médicos deberán informar a las pacientes en edad fértil acerca del riesgo potencial de la exposición a medicamentos que actúan sobre el sistema renina-angiotensina-aldosterona durante el embarazo. Cuando se detecta un embarazo, interrumpir la administración de la combinación amlodipino/valsartán lo antes posible. Rosuvastatina: No se ha establecido la seguridad de la rosuvastatina en mujeres embarazadas; por lo tanto, el uso de rosuvastatina está contraindicado durante el embarazo. Las mujeres en edad fértil deberán usar un método anticonceptivo efectivo. Debido a que el colesterol y otros productos de la biosíntesis del colesterol son esenciales para el desarrollo fetal, el beneficio del tratamiento durante el embarazo compensa el riesgo potencial de la inhibición de la HMG- CoA reductasa. Los estudios en animales proporcionan escasa evidencia sobre toxicidad reproductiva. Si una paciente queda embarazada durante el uso de este medicamento, se deberá interrumpir el tratamiento de inmediato. Lactancia materna: En estudios en animales, el valsartán y la rosuvastatina se excretan en la leche de ratas. El amlodipino se excreta en la leche materna. Por lo tanto, se deberá interrumpir el tratamiento con este fármaco durante la lactancia. Si el tratamiento es esencial, suspender la lactancia.
Reacciones adversas.
Combinación de amlodipino/valsartán/rosuvastatina: En el ensayo clínico se evaluó la seguridad de este medicamento en 203 pacientes con hipertensión y dislipidemia simultáneas; tratamiento de 8 semanas con la combinación amlodipino/ valsartán/ rosuvastatina (n=67), con la combinación amlodipino/valsartán (n=68), y con la combinación valsartán/rosuvastatina (n=68). A continuación, se presenta la lista de reacciones adversas informadas, independientemente de la relación con el fármaco de prueba, en el grupo con la combinación amlodipino/valsartán/rosuvastatina (n=67).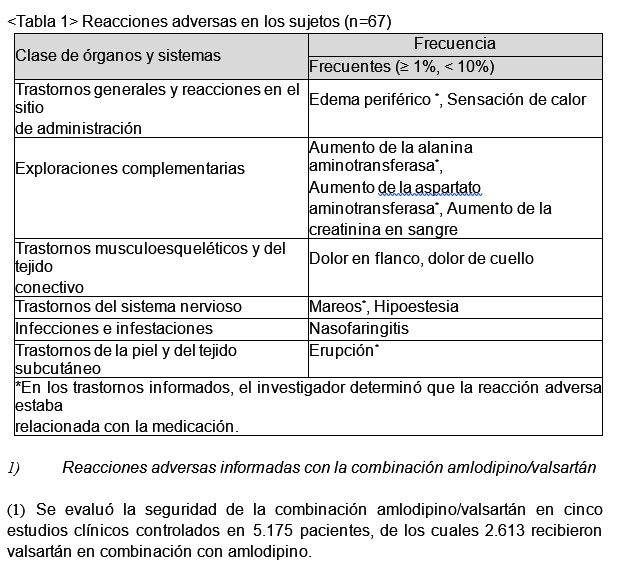
Las reacciones adversas se clasificaron por frecuencia utilizando la siguiente convención: muy frecuentes (≥1/10); frecuentes (≥1/100 a < 1/10); poco frecuentes (≥1/1.000 a < 1/100); raras (≥1/10.000 a < 1/1.000); muy raras, que incluye un solo caso informado ( < 1/10.000). Dentro del mismo grado de frecuencia, las reacciones se listan de mayor a menor gravedad.
En estudios clínicos controlados doble ciego, la incidencia de edema periférico, un efecto colateral reconocido del amlodipino en general fue menor en pacientes que recibieron la combinación amlodipino/valsartán (5,8%) que en los que recibieron amlodipino solo (9%). Resultado de la vigilancia post-comercialización. Como resultado de la vigilancia post-comercialización en 859 sujetos durante 6 años, que se realizó para reevaluación local, la tasa de reacciones adversas, independientemente de la relación con el fármaco de prueba, se informó en 5,12% (n=44, 56 casos). La tasa de las reacciones adversas para las cuales no se pudo excluir una relación con la combinación amlodipino/valsartán fue del 1,75% (n= 15, 19 casos). Entre ellas, la tasa para cefalea fue del 0,47% (n=4, 4 casos), para mareos del 0,35% (n=3, 3 casos), para prurito del 0,35% (n=3, 3 casos), para edema periférico del 0,23% (n=2, 2 casos), para hipotensión del 0,23% (n=2, 2 casos), y un caso cada uno de edema, dolor de pecho, dolor, tos y enuresis. No se informó ningún evento adverso grave, independientemente de la relación con la combinación amlodipino/valsartán. Se informaron 9 eventos adversos inesperados en total, que incluyeron molestia en el pecho en el 0,23% (n=2, 2 casos) y 1 caso cada uno de dolor, malestar abdominal, enfermedad por reflujo gastroesofágico, dolor de cuello, hipercolesterolemia, hipoglucemia, y enuresis. Se informaron 2 reacciones adversas inesperadas en total, que incluyeron 1 caso de dolor y otro de enuresis. Como resultado del análisis de los eventos adversos para reevaluación y el informe voluntario de eventos adversos locales con la base de datos de eventos adversos de todos los medicamentos comercializados, los eventos adversos informados con una frecuencia significativamente mayor desde el punto de vista estadístico con la combinación amlodipino/valsartán que con todos los demás fármacos son los listados a continuación Sin embargo, este resultado no significa que se demuestre la existencia de una relación entre los eventos adversos y las sustancias correspondientes. Trastornos generales: Dolor de pecho, aumento de peso. Sistema musculoesquelético: Artralgia, dolor musculoesquelético, rabdomiólisis. Cavidad oral: hiperplasia gingival. Aparato digestivo: Gastritis. Trastorno psiquiátrico: Nerviosismo. Sistema nervioso: Hipoestesia. Trastornos de la piel y del tejido subcutáneo: Síndrome de Stevens-Johnson. Reacciones adversas informadas con la combinación valsartán/rosuvastatina: Resultado de la vigilancia post-comercialización: El resultado del estudio de vigilancia post-comercialización en 698 sujetos durante 6 años para reevaluación, la tasa de eventos adversos, independientemente de la relación con el fármaco de prueba, fue del 14,90% (n=104/698, 137 casos). Entre ellos, los eventos adversos graves, independientemente de la relación, y las reacciones adversas al fármaco graves, para las cuales no se pudo excluir una relación con la medicación, se listan a continuación por orden de frecuencia.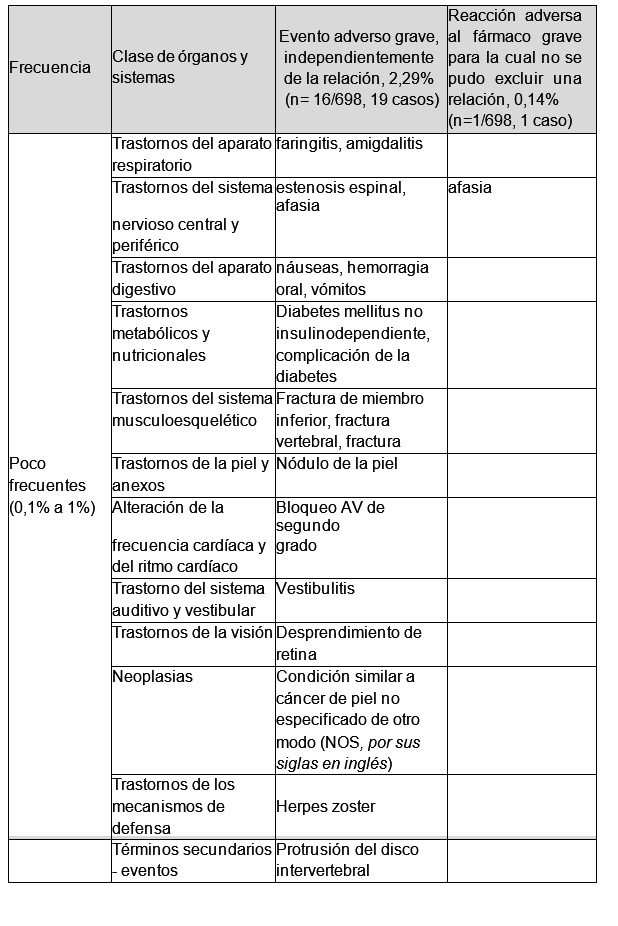
Y en la tabla siguiente se enumeran, en orden de frecuencia, los eventos adversos inesperados, independientemente de la relación, y las reacciones adversas al fármaco inesperadas para las cuales no se pudo excluir una relación con la medicación.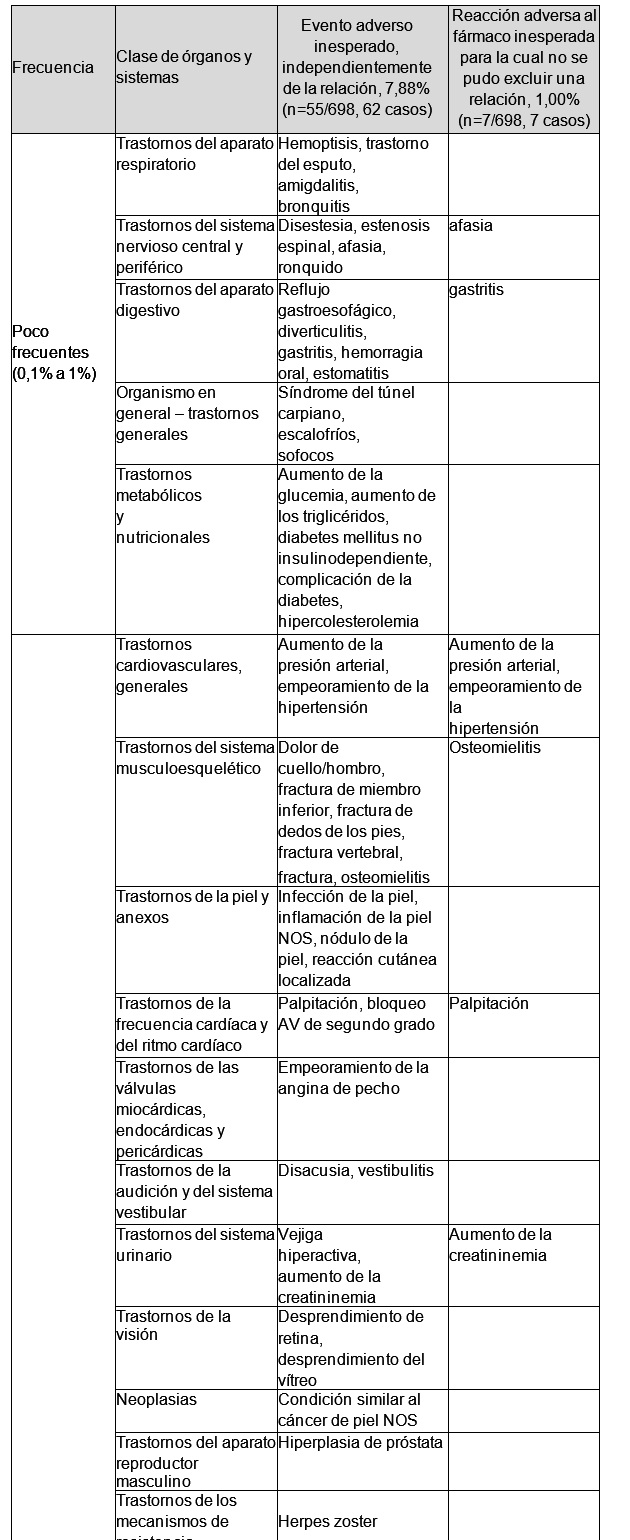
Información adicional de cada principio activo (monoterapia). Aunque no se observe un evento adverso en un ensayo clínico o en la vigilancia post- comercialización, es posible que se informe la aparición del alguno durante la monoterapia. Amlodipino
Las reacciones adversas, independientemente de la relación con la medicación, durante el estudio clínico de amlodipino como monoterapia se listan en la Tabla 3.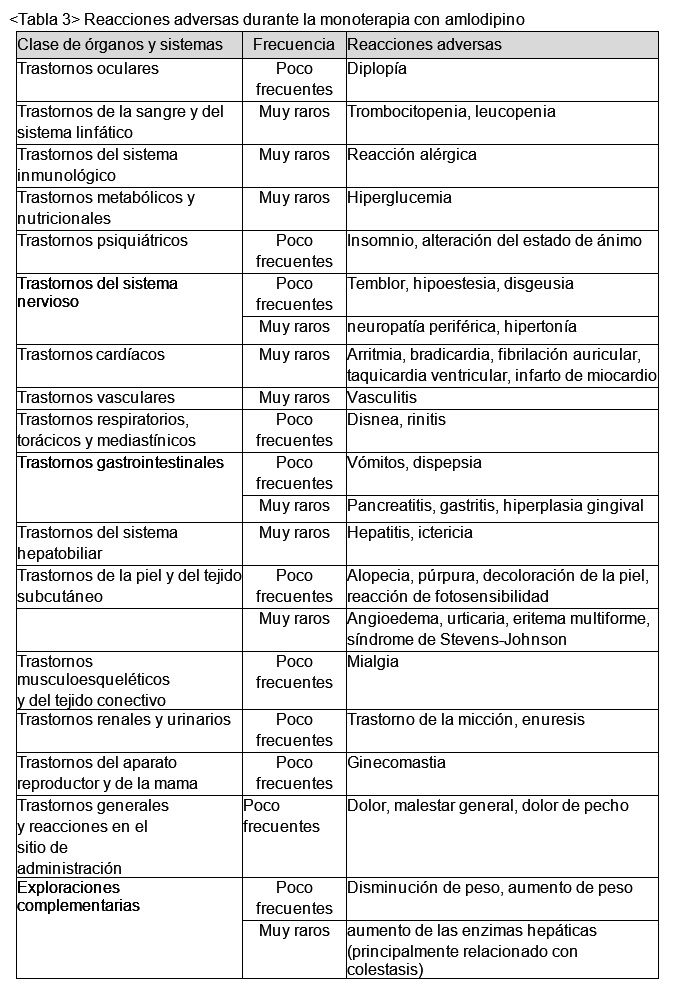
Se ha informado con poca frecuencia el siguiente evento post-comercialización donde una relación causal es incierta: ginecomastia. En la experiencia post-comercialización, se han informado ictericia y aumento de las enzimas hepáticas (principalmente compatibles con colestasis o hepatitis) en asociación con el uso de amlodipino, en algunos casos de gravedad suficiente para requerir hospitalización. Se informan leucopenia, trombocitopenia, estado de confusión, dermatitis exfoliativa, síndrome de Stevens-Johnson, reacción de fotosensibilidad, necrólisis epidérmica tóxica (desconocida). Para información de seguridad adicional, consultar la información autorizada de amlodipino como monoterapia.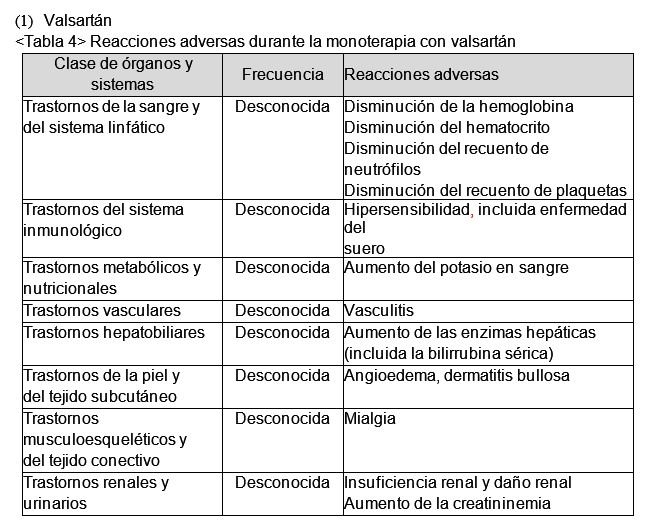
Los eventos adversos, que no puede determinarse si tienen una relación causal, ocurrieron en los estudios controlados de pacientes tratados con valsartán como monoterapia: insomnio, disminución de la libido, faringitis, rinitis, sinusitis, infección de las vías respiratorias superiores, infección viral. Para información de seguridad adicional, consultar la información autorizada de valsartán como monoterapia. Rosuvastatina: Las reacciones adversas observadas con rosuvastatina en general son leves y transitorias. En los estudios clínicos controlados, menos del 4% de los pacientes tratados con rosuvastatina fueron retirados debido a reacciones adversas. La frecuencia de las reacciones adversas se clasifica de acuerdo con la siguiente convención: Frecuentes (≥1/100 a < 1/10); Poco frecuentes (≥1/1.000 a < 1/100); Raros (≥1/10.000 a < 1/1000); Muy raros ( < 1/10.000); Desconocida (no puede estimarse de los datos disponibles).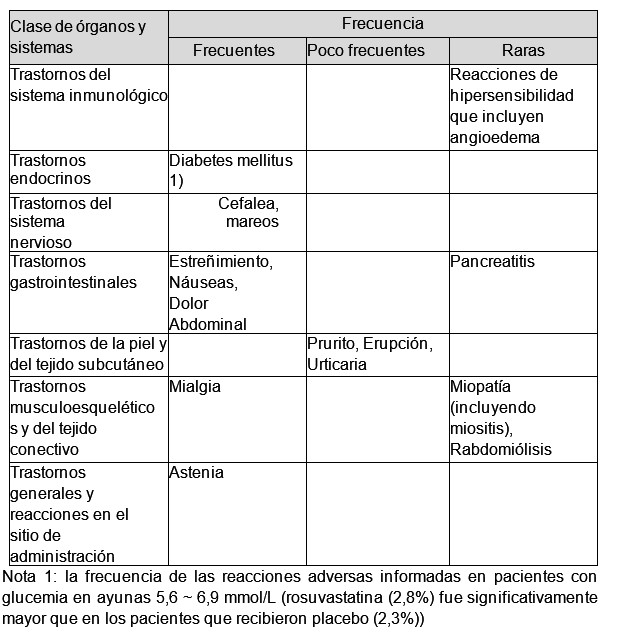
Al igual que con otros inhibidores de la HMG-CoA reductasa, la incidencia de reacciones adversas tiende a ser dependiente de la dosis. Efectos renales: Se observó proteinuria, detectada mediante prueba con tira reactiva y principalmente de origen tubular. Se observaron variaciones de la proteinuria, de inexistente o trazas a ++ o más, en < 1% de los pacientes en algún momento durante el tratamiento con 10 y 20 mg, y en aproximadamente el 3% de los pacientes tratados con 40 mg. Se observó un aumento menor en el cambio de inexistente o trazas a + con la dosis de 20 mg. En la mayoría de los casos, la proteinuria disminuye o desaparece espontáneamente con el tratamiento continuo. No se ha demostrado que la proteinuria sea predictiva de enfermedad renal aguda o progresiva. Se observó hematuria en pacientes tratados con rosuvastatina y los datos de los ensayos clínicos revelan que su incidencia es baja. Efectos musculoesqueléticos: Se informaron efectos sobre el músculo esquelético por ej., mialgia, miopatía (incluida miositis) y, rara vez, rabdomiólisis con y sin insuficiencia renal aguda en los pacientes tratados con rosuvastatina con todas las dosis y en particular con dosis > 20 mg. Se observó un aumento relacionado con la dosis en los niveles de la creatinquinasa (CK) en pacientes que tomaban rosuvastatina; la mayoría de los casos fueron leves, asintomáticos y transitorios. Si los niveles de CK están marcadamente elevados ( > 5xLSN), se deberá interrumpir el tratamiento. Efectos hepáticos: Al igual que con otros inhibidores de la HMG-CoA reductasa, se observó un aumento de las transaminasas relacionado con la dosis en un pequeño número de pacientes que tomaban rosuvastatina; la mayoría de los casos fueron leves, asintomáticos y transitorios. Experiencia post-comercialización internacional: Además de las reacciones adversas mencionadas, se informaron las siguientes reacciones adversas. Sistema nervioso: muy raramente polineuropatía, amnesia, neuropatía periférica (desconocida). Aparato respiratorio y tórax: tos, disnea (desconocida). Aparato digestivo: diarrea (desconocida).Trastorno hematológico: trombocitopenia (desconocida). Hepatobiliares: Muy raramente ictericia, hepatitis, raramente aumento de las transaminasas. Trastornos de la piel y del tejido subcutáneo: Síndrome de Stevens-Johnson (desconocida), reacción al fármaco con eosinofilia y síntomas sistémicos (desconocida). Sistema musculoesquelético: raramente síndrome similar al lupus, desgarro muscular, muy raramente artralgia, miopatía necrotizante inmunomediada (desconocida). Trastorno renal: Muy raramente hematuria. Otras: edema (desconocida). Se informaron las siguientes reacciones adversas a algunas de las estatinas: Sistema psiconeurológico: depresión, trastorno del sueño (incluidos insomnio y pesadillas) (desconocida). Aparato respiratorio: excepcionalmente enfermedad pulmonar intersticial con la administración prolongada Aparato genitourinario: disfunción sexual, ginecomastia (desconocida). Hepatobiliares: insuficiencia hepática fatal y no fatal. Hubo informes post-comercialización excepcionales de trastorno cognitivo (por ej., hipoamnesia, olvido, amnesia, deterioro de la memoria, y confusión) asociado con el uso de estatinas. Estos problemas cognitivos se informaron para todas las estatinas. En general, los informes no son graves y se revierten al interrumpir el uso de estatinas, con tiempos variables hasta el comienzo de los síntomas (1 día a años) y la resolución de los síntomas (mediana de 3 semanas). Resultado de la vigilancia post-comercialización local: En la vigilancia post-comercialización para reevaluación, que se realizó en 3.081 sujetos durante 6 años, la tasa de incidencia de eventos adversos inesperados, independientemente de la relación con la medicación, fue del 10,06% (310 sujetos, 415 casos). En su mayoría fueron cefalea 0,78% (n=24, 24 casos), mareos 0,75% (n=23, 23 casos), aumento de la alanina aminotransferasa 0,58% (n=18, 18 casos), dolor de pecho, tos, mialgia 0,49% cada uno (n=15, 15 casos). La tasa de reacciones adversas al fármaco inesperadas, para las que no pudo excluirse una relación con la rosuvastatina, fue del 2,92% (n=90, 106 casos). En las reacciones adversas al fármaco informadas, se observó principalmente aumento de la alanina aminotransferasa en el 0,55% (n=17, 17 casos). Y se observaron otras reacciones adversas al fármaco en este orden; mialgia 0,42% (n=13, 13 casos); cefalea 0,39% (n=12, 12 casos); aumento de la creatinfosfoquinasa en sangre 0,29% (n=9, 9 casos); mareos 0,26% (n=8, 8 casos); estreñimiento, aumento de la aspartato aminotransferasa 0,16% cada uno (n=5, 5 casos); astenia, artralgia 0,13% cada uno (n=4, 4 casos); fatiga, anestesia 0,10% cada uno (n=3, 3 casos); disestesia, molestia en el pecho, náuseas, dolor abdominal, diarrea, disminución del apetito, distensión abdominal, prurito, prueba de la función hepática anormal 0,06% cada uno (n=2, 2 casos); síncope, pantalgia, calambre, gota, disfunción eréctil 0,03% cada uno (n=1, 1 caso). En un sujeto se observaron mialgia y artralgia que fueron reacciones adversas al fármaco graves. Las reacciones adversas inesperadas y no informadas antes de la comercialización fueron las siguientes: artralgia 0,13% (n=4, 4 casos); fatiga, anestesia 0,10% cada uno (n=3, 3 casos); disestesia, molestia en el pecho, disminución del apetito, distensión abdominal, prueba de la función hepática anormal 0,06% cada uno (n=2, 2 casos); síncope, pantalgia, calambre, gota, disfunción eréctil 0,03% cada uno (n=1, 1 caso). Además, se informó 1 caso de artralgia como reacción adversa al fármaco grave inesperada. Se informaron 98 reacciones adversas espontáneamente durante el período de reevaluación. Entre los casos antes mencionados, 2 casos de insuficiencia renal aguda y 1 caso cada uno de oliguria, trombocitopenia, y aumento de la creatinina en sangre fueron reacciones adversas al fármaco graves inesperadas.
Precauciones.
Conducción de vehículos y uso de maquinaria: No se han realizado estudios para determinar el efecto sobre la capacidad para conducir y usar maquinaria. Los pacientes deberán tener en cuenta que ocasionalmente pueden aparecer mareos o cansancio. Al igual que otros antihipertensivos, se recomienda cuidado especialmente al conducir o usar maquinaria. Amlodipino: Uso en pacientes con insuficiencia cardíaca: En un estudio de seguimiento a largo plazo controlado con placebo (PRAISE-2) de amlodipino en pacientes con insuficiencia cardíaca NYHA III y IV sin síntomas clínicos ni hallazgos objetivos indicadores de enfermedad isquémica subyacente, el amlodipino no tuvo efecto sobre la mortalidad cardiovascular total. En esta misma población, el amlodipino estuvo asociado con un aumento de los informes de edema pulmonar. Pacientes con insuficiencia hepática: Los pacientes con insuficiencia hepática tienen una disminución de la depuración de amlodipino resultante en una mayor vida media. No se han establecido recomendaciones de dosis en estos pacientes; por consiguiente, se debe elegir la dosis con cuidado. Después de la discontinuación de este medicamento, se observa un efecto hipotensor prolongado debido a la larga vida media en plasma. Por lo tanto, el reemplazo del tratamiento requiere un cuidadoso seguimiento y ajuste de la dosis/del intervalo del tratamiento de reemplazo. Debido al lento comienzo de acción, no deberían esperarse efectos en pacientes con angina inestable, que requieren tratamiento de emergencia. Valsartán: Puede producirse hipotensión excesiva transitoria (con síncope, pérdida de conocimiento, etc.) en el paciente el primer día de uso de este medicamento. En este caso, interrumpir el uso del medicamento y tratar al paciente apropiadamente. Para los pacientes que inician el tratamiento con una dosis baja, controlar cuidadosamente y aumentar la dosis en forma gradual, especialmente en los siguientes casos. Paciente en hemodiálisis: Paciente que usa diuréticos (en pacientes con depleción grave de sodio y/o volumen, rara vez puede ocurrir hipotensión sintomática.) Paciente con dieta hiposódica: En pacientes con depleción grave de sodio y/o volumen debido a diuréticos en dosis altas, rara vez puede ocurrir hipotensión sintomática. Se recomienda ajustar la depleción de sodio y/o volumen reduciendo las dosis de diuréticos o con otros métodos antes de usar valsartán. Si se produce hipotensión, colocar al paciente en posición supina y, si fuera necesario, administrar solución salina intravenosa mediante instilación. Continuar con el tratamiento una vez que la presión arterial se haya estabilizado. Pacientes con estenosis de la arteria renal: La administración a corto plazo de valsartán a doce pacientes con hipertensión renovascular secundaria a estenosis de la arteria renal unilateral no produjo cambios significativos en la hemodinamia renal, la creatinina sérica, ni el nitrógeno ureico en sangre (BUN, por sus siglas en inglés). Sin embargo, otros fármacos que afectan el sistema renina-angiotensina-aldosterona pueden aumentar la urea en sangre y la creatinina sérica en pacientes con estenosis de la arteria renal unilateral o bilateral, por lo tanto, se recomienda controlar la función renal cuando los pacientes reciben tratamiento con valsartán. En pacientes con estenosis de la arteria renal bilateral o estenosis de un riñón solitario, se observó insuficiencia renal aguda inducida por disminución del flujo renal y de la presión de filtración glomerular. La dosis de valsartán no deberá exceder los 80 mg en pacientes con insuficiencia hepática leve a moderada. Casi todo el valsartán absorbido se elimina en la bilis, esencialmente sin cambios, y se observó una baja tasa de eliminación en pacientes con obstrucción biliar. Por lo tanto, se deberá evitar el uso de este fármaco en dichos pacientes. Se observó duplicación de la exposición (AUC) en pacientes con insuficiencia hepática leve a moderada en comparación con sujeto sanos. En pacientes post-infarto de miocardio y/o insuficiencia cardíaca que usan valsartán, la presión arterial disminuye en la mayoría de los casos, pero no es necesaria la interrupción del tratamiento si la hipotensión persiste. Es necesario tener cuidado al iniciar el uso de valsartán en pacientes post-infarto de miocardio y/o insuficiencia cardíaca. Como resultado de la inhibición del sistema renina-angiotensina-aldosterona (SRAA), se espera un cambio en la función renal. En pacientes cuya función renal puede depender de la actividad del sistema renina-angiotensina-aldosterona (por ej. pacientes con insuficiencia cardíaca congestiva grave), el tratamiento con inhibidores de la ECA y con antagonistas de la angiotensina II ha estado asociado con oliguria y/o azoemia progresiva y rara vez con insuficiencia renal aguda y/o muerte. Se observaron síntomas similares en pacientes que usan valsartán, por lo tanto, no puede excluirse que el uso de valsartán pueda estar asociado con deterioro de la función renal. Tampoco se recomienda la triple combinación de un inhibidor de la ECA, un antagonista de los receptores de mineralocorticoides, y valsartán debido a que esta combinación aparentemente aumenta el riesgo de morbimortalidad por insuficiencia cardíaca. En algunos pacientes con insuficiencia cardíaca, se observó aumento de los niveles de nitrógeno ureico en sangre (BUN), creatinina, y potasio. En general, este síntoma es menor y transitorio. Es posible que este síntoma se observe más en pacientes con deterioro de la función renal. Se deberá considerar la interrupción o reducción de la dosis de valsartán y/o de los diuréticos. Dado que el nivel de potasio sérico puede aumentar en pacientes con hiperpotasemia se deberá evitar el uso de este medicamento a menos que el tratamiento se considere esencial. Es posible que se observe hipopotasemia en pacientes con insuficiencia renal, diabetes, y otros pacientes en los que los niveles de potasio sérico aumentan con facilidad. Se deberá controlar cuidadosamente el nivel de potasio sérico. Actualmente no hay experiencia sobre el uso seguro de valsartán en pacientes con trasplante renal reciente. Rosuvastatina: Enfermedad pulmonar intersticial: Se han informado casos excepcionales de enfermedad pulmonar intersticial con algunas estatinas, especialmente con el tratamiento a largo plazo. Las características de presentación pueden incluir disnea, tos no productiva y deterioro de la salud general (fatiga, pérdida de peso y fiebre). Si se sospecha que un paciente ha desarrollado enfermedad pulmonar intersticial, se deberá interrumpir el tratamiento con estatinas. Hubo informes post-comercialización excepcionales de insuficiencia hepática fatal y no fatal en pacientes que tomaban estatinas, incluida la rosuvastatina. Si durante el tratamiento con rosuvastatina se produce daño hepático grave con síntomas clínicos y/o hiperbilirrubinemia o ictericia, interrumpir de inmediato el tratamiento. Si no se establece otra etiología, no reanudar este fármaco. Se han informado aumentos en los niveles de HbA1c y glucosa sérica en ayunas con los inhibidores de la HMG-CoA reductasa, incluida la rosuvastatina. Diabetes mellitus: Algunas evidencias indican que las estatinas como clase aumentan la glucemia y en algunos pacientes, con alto riesgo de diabetes en el futuro, pueden producir un nivel de hiperglucemia donde es apropiado un cuidado formal de la diabetes. Sin embargo, este riesgo se compensa con la reducción del riesgo vascular con estatinas y, por lo tanto, no deberá ser una razón para interrumpir el tratamiento con estatinas. Los pacientes en riesgo (glucosa en ayunas 5,6 a 6,9 mmol/L, IMC > 30 kg/m2, triglicéridos elevados, hipertensión) deberán ser controlados tanto clínica como bioquímicamente de acuerdo con las guías de tratamiento. En el estudio JUPITER, la frecuencia general informada de diabetes mellitus fue del 2,8% en el grupo con rosuvastatina y del 2,3% en el grupo con placebo, principalmente en pacientes con glucosa en ayunas de 5,6 a 6,9 mmol/L. Polimorfismos genéticos: Se informa que los polimorfismos individuales de SLCO1B1(OATP1B1) c.521CC y ABCG2(BCRP) c.421AA están asociados con una mayor exposición a rosuvastatina (AUC) en comparación con los genotipos SLCO1B1 c.521TT o ABCG2 c.421CC. Esta genotipificación específica no está establecida en la práctica clínica, pero para los pacientes que se sabe que tienen estos tipos de polimorfismos, se recomienda una dosis diaria más baja de rosuvastatina. Miopatía necrotizante inmunomediada (MNIM): Se informaron casos de MNIM, una enfermedad muscular autoinmune asociada con el uso de estatinas. La MNIM se caracteriza por: debilidad de los músculos proximales y creatinquinasa sérica elevada, las cuales persisten a pesar de la discontinuación del tratamiento con estatinas; biopsia muscular que demuestra miopatía necrotizante; y mejoría con inmunosupresores. Reacciones adversas cutáneas graves: Se informaron reacciones adversas cutáneas graves fatales como Síndrome de Stevens-Johnson (SSJ) y reacción al fármaco con eosinofilia y síntomas