EPIVIR® 3TC
GLAXOSMITHKLINE
Comprimidos recubiertos
Antiviral.
Composición.
La formulación EPIVIR® en comprimidos recubiertos contiene 150 mg ó 300 mg de lamivudina. Los comprimidos de 150 mg son blancos, ranurados, en forma de diamante y se encuentran marcados con el código "GX CJ7" en ambas caras. Presentación farmacéutica: Comprimidos Recubiertos. Lista de Excipientes: Comprimidos: Núcleo del comprimido: celulosa microcristalina, glicolato sódico de almidón Tipo A, estearato de magnesio. Película de recubrimiento de los comprimidos blancos: hidroxipropilmetilcelulosa, dióxido de titanio, óxido de hierro negro (E172) (sólo las comprimidos de 300mg), macrogol, polisorbato 80.
Indicaciones.
La formulación EPIVIR®, en combinación con otros agentes antirretrovíricos, se indica en el tratamiento de adultos y niños infectados por el VIH.
Dosificación.
La terapia con EPIVIR® debe ser iniciada por un médico con experiencia en el tratamiento de la infección ocasionada por VIH. EPIVIR® puede tomarse con o sin alimentos. Para garantizar la administración de la dosis completa, sería ideal que lo(s) comprimidos(s) fueran(n) deglutidos sin triturarse. Para aquellos pacientes que no puedan deglutir comprimidos, lamivudina se encuentra disponible en solución oral. Alternativamente los comprimidos pueden ser triturados y adicionados a una pequeña cantidad de líquido o alimento semisólido, que deberá consumirse inmediatamente (ver Farmacocinética). Adultos y adolescentes con un peso corporal de cuando menos 30 kg: La dosis recomendada de EPIVIR® consiste en 300 mg al día. Ésta puede administrarse como 150 mg (15 mL de solución oral, 1 comprimido de 150 mg), dos veces al día, ó 300 mg (30 mL de solución oral, 2 comprimidos de 150 mg ó 1 comprimido de 300 mg) una vez al día. Niños mayores de tres meses de edad y con un peso corporal menor de 30 kg. Solución oral: La dosis recomendada consiste en 4 mg/kg, administrados dos veces al día, hasta un máximo de 300 mg al día. Comprimidos: Niños con un peso corporal entre 21 kg y 30 kg: La dosis oral recomendada de EPIVIR® (150 mg) consiste en medio comprimido tomado en la mañana y un comprimido completo tomado en la noche. Niños con un peso corporal de 14 kg a 21 kg: La dosis oral recomendada de EPIVIR® (150 mg) consiste en medio comprimido ranurado tomado dos veces al día. Niños menores de tres meses de edad: Los pocos datos disponibles resultan insuficientes para establecer recomendaciones específicas de dosificación (ver Farmacocinética). Pacientes de edad avanzada: No se dispone de datos específicos, sin embargo, se recomienda tener un cuidado especial en este grupo de edad debido a los cambios asociados con la edad, como la disminución en la función renal y la alteración de los parámetros hematológicos. Insuficiencia renal: Las concentraciones plasmáticas de lamivudina (ABC) aumentan en los pacientes que exhiben insuficiencia renal de grado moderado a severo, debido a la disminución en la depuración (ver Farmacocinética). Por tanto, la dosis debe reducirse en aquellos pacientes con una depuración de creatinina inferior a 50 mL/min, de acuerdo con la siguiente tabla. Los mismos porcentajes de reducción en la dosificación aplican para los pacientes pediátricos con insuficiencia renal. Cuando se requiere la administración de dosis inferiores a 150 mg, se recomienda emplear la formulación EPIVIR® en solución oral.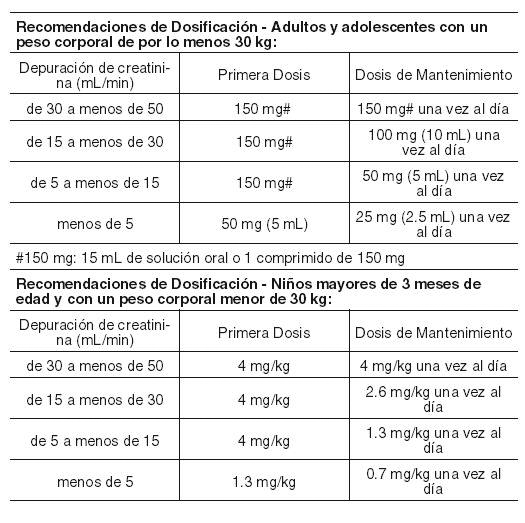
Insuficiencia hepática: No se requiere realizar ajustes en la dosificación de los pacientes con insuficiencia hepática de grado moderado o severo, a menos que el paciente también exhiba insuficiencia renal (ver Farmacocinética).
Contraindicaciones.
El uso de EPIVIR® se contraindica en los pacientes con hipersensibilidad conocida a la lamivudina o a cualquier ingrediente de la preparación.
Reacciones adversas.
Los eventos que se listan a continuación se han comunicado durante el tratamiento de la enfermedad ocasionada por el VIH, con EPIVIR® administrado solo y en combinación con otros agentes antirretrovíricos. En muchos casos, no es claro si esos eventos se relacionan con los medicamentos o si son el resultado del proceso patológico subyacente. Se ha empleado la siguiente convención para la clasificación de los efectos adversos: muy común ( > 1/10), común ( > 1/100, < 1/10), no común ( > 1/1,000, < 1/100), raro ( > 1/10,000, < 1/1,000) muy raro ( < 1/10,000). Trastornos sanguíneos y del sistema linfático: No comunes: Neutropenia, anemia, trombocitopenia. Muy raro: Aplasia eritrocítica pura. Trastornos metabólicos y nutricionales: Común: Hiperlactatemia. Raro: Acidosis láctica (ver Advertencias y Precauciones). Redistribución/acumulación de grasa corporal (ver Advertencias y Precauciones). La incidencia de ocurrencia de este evento depende de diversos factores, incluyendo la combinación particular de fármacos antirretrovíricos. Trastornos del sistema nervioso: Común: Cefalea. Insomnio. Muy raros: Parestesia. Se han comunicado casos de neuropatía periférica, aunque no se sabe con precisión si existe alguna relación causal con el tratamiento. Trastornos respiratorios: Común: Tos, síntomas nasales. Trastornos gastrointestinales: Comunes: Náuseas, vómito, dolor en el abdomen superior, diarrea. Raros: Pancreatitis, aunque no se sabe con precisión si existe alguna relación causal con el tratamiento. Aumentos en los niveles séricos de amilasa. Trastornos hepatobiliares: No comunes: Elevaciones transitorias en los niveles de enzimas hepáticas (ASAT, ALAT). Trastornos de la piel y del tejido subcutáneo: Comunes: Exantema, alopecia. Trastornos musculoesqueléticos y del tejido conjuntivo: Comunes: Artralgia, trastornos musculares. Raro: Rabdomiólisis. Trastornos generales y en el sitio de administración: Comunes: Fatiga, malestar general, fiebre.
Advertencias.
No se recomienda el uso de la formulación EPIVIR® como monoterapia. Se debe advertir a los pacientes que aún no se demuestra que la terapia antirretrovírica actual, incluyendo EPIVIR®, evite el riesgo de transmitir el VIH a otras personas a través del contacto sexual o por contaminación sanguínea. Por tanto, se deben seguir tomando las precauciones adecuadas. Existe la posibilidad de que los pacientes tratados con EPIVIR®, o con cualquier otra terapia antirretrovírica, sigan desarrollando infecciones oportunistas y otras complicaciones de la infección por VIH, por lo que deben permanecer bajo estrecha observación clínica por médicos con experiencia en el tratamiento de pacientes con enfermedades asociadas con el VIH. Insuficiencia renal: Las concentraciones plasmáticas de lamivudina (ABC) aumentan en los pacientes con insuficiencia renal de grado moderado a severo, debido a la disminución en su depuración. Por tanto, debe realizarse un ajuste en la dosificación (ver Dosis y Administración). Pancreatitis: Se ha observado pancreatitis en algunos pacientes que reciben EPIVIR®. Sin embargo, no es claro si este trastorno se debió al tratamiento medicamentoso o a la enfermedad por VIH ya existente. Se debe considerar la posibilidad de ocurrencia de pancreatitis cada vez que un paciente desarrolle dolor abdominal, náuseas, vómito o se le detecten marcadores bioquímicos elevados. Se debe suspender la administración de EPIVIR® hasta descartar un diagnóstico de pancreatitis. Acidosis láctica/hepatomegalia severa con esteatosis: Se han comunicado casos de acidosis láctica y hepatomegalia severa con esteatosis, con inclusión de casos mortales, al usar análogos de nucleósido antirretrovíricos, ya sea solos o en combinación, incluyendo lamivudina. La mayoría de estos casos ha tenido lugar en mujeres. Las manifestaciones clínicas que pueden indicar el desarrollo de acidosis láctica incluyen debilidad generalizada, anorexia y pérdida de peso súbita e inexplicable, así como síntomas gastrointestinales y síntomas respiratorios (disnea y taquipnea). Se debe tener precaución al administrar EPIVIR® a cualquier paciente, particularmente a los que se sabe exhiben factores de riesgo de padecer enfermedades hepáticas. Se debe suspender el tratamiento con EPIVIR® en cualquier paciente que desarrolle hallazgos clínicos o de laboratorio que sugieran la presencia de acidosis láctica o hepatotoxicidad (que pueden incluir hepatomegalia y esteatosis, aún en ausencia de elevaciones muy notables en los niveles de aminotransferasas). Los pacientes con hepatitis C concomitante y tratados con interferón alfa y ribavirina pueden constituir un grupo de riesgo especial. Redistribución de la grasa: En algunos pacientes que reciben terapia antirretrovírica de combinación, se ha observado una redistribución / acumulación de grasa corporal, con inclusión de obesidad central, aumento de la grasa dorsocervical (joroba de búfalo), disminución de la grasa periférica, disminución de la grasa facial, crecimiento mamario, niveles elevados de lípidos séricos y glucosa sanguínea, ya sea por separado o conjuntamente (ver Efectos Adversos). Aunque se ha asociado a todos los miembros de las clases de medicamentos PI (inhibidores de la proteasa) y NRTI (inhibidores nucleosídicos de la transcriptasa inversa), con uno o más de estos efectos adversos específicos, ligados a un síndrome general conocido comúnmente como lipodistrofia, los datos disponibles indican que existen diferencias en cuanto al riesgo entre los miembros individuales de las respectivas clases terapéuticas. Además, el síndrome de lipodistrofia tiene una etiología multifactorial; por ejemplo, con el estado de la enfermedad ocasionada por el VIH, la edad avanzada y la duración del tratamiento antirretrovírico, donde todos desempeñan papeles importantes, posiblemente sinérgicos. En la actualidad, se desconocen las consecuencias a largo plazo de estos efectos. El examen clínico debe incluir una evaluación de los signos físicos de la redistribución de grasa. Se debe considerar la medición de los niveles de lípidos séricos y glucosa sanguínea. Los trastornos lipídicos deben tratarse según sea clínicamente adecuado. Síndrome de Reconstitución Inmunológica: En aquellos pacientes infectados por el VIH, que presentan una deficiencia inmunitaria de grado severo al momento de iniciar la terapia antirretrovírica (TAR), puede ocurrir una reacción inflamatoria, o infecciones oportunistas asintomáticas o residuales, que ocasionen serios trastornos clínicos o un agravamiento de los síntomas. Normalmente estas reacciones se observan dentro de las primeras semanas o meses posteriores a la iniciación de la TAR. Ejemplos importantes son la retinitis citomegalovírica, infecciones micobacterianas generalizadas o focales, o ambas, así como neumonía ocasionada por cepas de Pneumocystis jiroveci (P. carinii). Se debe evaluar, sin demora alguna, cualquier síntoma inflamatorio que se presente y, cuando sea necesario, iniciar un tratamiento. Pacientes coinfectados por el virus de Hepatitis B: Las pruebas clínicas, así como el uso comercial de la formulación EPIVIR®, han mostrado que algunos pacientes que padecen enfermedad crónica ocasionada por el virus de hepatitis B (VHB) pueden experimentar indicios clínicos, o de laboratorio, de hepatitis recurrente a la suspensión de la terapia con EPIVIR®, lo cual podría tener consecuencias más severas en aquellos pacientes con enfermedad hepática descompensada. Si se suspende la administración de EPIVIR® en algún paciente que presente una coinfección por VIH y VHB, debe considerarse una vigilancia periódica tanto de las pruebas de función hepática como de los marcadores de replicación del VHB. Solución oral: Se debe advertir a los pacientes diabéticos que cada dosis para adultos contiene 3 g de sacarosa.
Interacciones.
La probabilidad de que se presenten interacciones es baja, ya que tanto la fijación a proteínas plasmáticas como el metabolismo son limitados y, además, casi toda la lamivudina se elimina por la vía renal en forma inalterada. La lamivudina se elimina principalmente por secreción catiónica orgánica activa. Se debe considerar la posibilidad de que se presenten interacciones con otros medicamentos administrados concurrentemente, en particular cuando su principal ruta de eliminación sea la secreción renal activa por medio del sistema de transporte catiónico orgánico, p.ej., el trimetoprim. Otras sustancias activas (p.ej., ranitidina y cimetidina) sólo se eliminan parcialmente a través de este mecanismo, demostrándose que no interactúan con la lamivudina. No es probable que las sustancias activas que se excretan principalmente por la vía aniónica orgánica activa, o por filtración glomerular, produzcan interacciones clínicamente significativas con la lamivudina. Zidovudina: Se observó una modesta elevación en la Cmáx (28%) de zidovudina cuando se administró con lamivudina; sin embargo, la exposición global (ABC) no se vio alterada en forma significativa. La zidovudina no tuvo efecto alguno sobre la farmacocinética de la lamivudina (ver Farmacocinética). Trimetoprim/sulfametoxazol: La administración de 160 mg/800 mg de trimetoprim/sulfametoxazol (cotrimoxazol) provoca un aumento del 40% en la exposición a la lamivudina, debido al componente trimetoprim. Sin embargo, a menos que el paciente padezca insuficiencia renal, no es necesario realizar ajustes en la dosificación de EPIVIR® (ver Dosis y Administración). La lamivudina no tiene efecto alguno sobre la farmacocinética del trimetoprim o del sulfametoxazol. No se ha estudiado el efecto resultante de la coadministración de EPIVIR®, con dosis elevadas de cotrimoxazol, para el tratamiento de toxoplasmosis y neumonía ocasionada por cepas de Pneumocystis jiroveci (P. carinii). Zalcitabina: Existe la posibilidad de que la lamivudina inhiba la fosforilación intracelular de la zalcitabina, cuando los dos medicamentos se emplean concurrentemente. Por tanto, no se recomienda el uso de EPIVIR® en combinación con zalcitabina. Didanosina: dado que su eliminación no ocurre por el mecanismo propio de lamivudina, es improbable que interactúe con esta última. Embarazo y Lactancia: Embarazo: Existen pocos datos disponibles sobre el uso seguro de EPIVIR® durante el embarazo humano. Los estudios realizados en humanos han confirmado que la lamivudina atraviesa la placenta. Su uso durante el embarazo sólo debe considerarse si el beneficio excede el riesgo. Aunque los resultados de los estudios realizados en animales (ver Datos Preclínicos de Seguridad) no siempre sirven para pronosticar la respuesta humana, los hallazgos en conejos sugieren un riesgo potencial de pérdida embrionaria temprana. Aunque los estudios de reproducción con animales no siempre predicen la respuesta en humanos, no se recomienda la administración durante los tres primeros meses de embarazo. Se han producido comunicaciones de elevaciones transitorias leves en los niveles séricos de lactato, las cuales pueden deberse a una disfunción mitocondrial, en neonatos y lactantes expuestos, in utero o periparto, a inhibidores nucleosídicos de la transcriptasa inversa (NRTI, por sus siglas en inglés). Se desconoce la importancia clínica de las elevaciones transitorias en los niveles séricos de lactato. En muy raras ocasiones, también han surgido comunicaciones de retraso en el desarrollo, convulsiones y otras enfermedades neurológicas. Sin embargo, no se ha establecido relación causal alguna entre estos eventos y la exposición a NRTI, in utero o periparto. Estos hallazgos no afectan las recomendaciones actuales concernientes al uso de terapia antirretrovírica en mujeres embarazadas para prevenir la transmisión vertical de VIH. Lactancia: Los expertos en salud recomiendan que, siempre que sea posible, las mujeres infectadas con VIH no amamanten a sus bebés con el fin de evitar la transmisión del VIH. Después de su administración oral, la lamivudina se excretó en la leche materna a concentraciones similares a las encontradas en el suero (1 a 8 microgramos/mL). Como la lamivudina y el virus alcanzan la leche materna, es recomendable que las madres que reciben EPIVIR® no amamanten a sus bebés. Efectos sobre la Capacidad de Conducir y Operar Maquinaria: No se han realizado estudios para investigar el efecto de EPIVIR® sobre la capacidad de conducir vehículos u operar maquinaria. Además, con base en la farmacología de la lamivudina, no puede predecirse efecto nocivo alguno en estas actividades. No obstante, al considerar la capacidad del paciente para conducir vehículos u operar maquinaria, debe tenerse presente su estado clínico y el perfil de efectos adversos de EPIVIR®.
Incompatibilidades.
Ninguna comunicada.
Conservación.
No almacenar la formulación EPIVIR® en comprimidos a temperaturas superiores a 30°C. Vida de Anaquel: La fecha de caducidad se indica en el empaque.
Sobredosificación.
Existen pocos datos disponibles sobre las consecuencias de la ingestión de sobredosis agudas en humanos. No hubo fallecimientos y los pacientes se recuperaron. No se han identificado síntomas o signos específicos posteriores a la sobredosis. En caso de sobredosificación, debe vigilarse al paciente y aplicarse el tratamiento estándar de soporte que sea necesario. Como la lamivudina puede eliminarse por diálisis, se puede emplear hemodiálisis continua para tratar una sobredosificación, aunque esto no se ha estudiado.
Presentación.
Comprimidos: Cajas de cartón que contienen comprimidos dentro de un frasco blanco de polietileno de alta densidad (HDPE), con cierre a prueba de niños. Comprimidos x 60.
EPIVIR® 3TC
GLAXOSMITHKLINE
Solución oral
Antiviral.
Composición.
La formulación EPIVIR® en solución oral contiene 10 mg/mL de lamivudina, en una solución que contiene sacarosa al 20% (p/v). Es una solución transparente, cuya tonalidad va de incolora a amarillo claro. Presentación farmacéutica: Solución Oral. Lista de Excipientes: Solución oral: Sacarosa (al 20% p/v), Hidroxibenzoato de metilo, Hidroxibenzoato de propilo, Acido cítrico anhidro, Propilenglicol, Citrato de sodio, Sabor artificial a fresa, Sabor artificial a plátano, Agua (purificada).
Farmacología.
Farmacodinamia: Grupo farmacoterapéutico: análogo de nucleósido; Código ATC: J05AF05. La lamivudina es un potente inhibidor selectivo de la replicación de VIH-1 y VIH-2 in vitro. También es activa contra aislados clínicos de VIH resistentes a la zidovudina. La lamivudina se metaboliza intracelularmente para formar el 5'-trifosfato, la entidad activa, la cual tiene una vida media intracelular de 16 a 19 horas. El 5'-trifosfato de lamivudina es un inhibidor débil de las actividades de la transcriptasa inversa del VIH dependientes de ARN y ADN; su principal modo de acción es como terminador de la cadena de la transcripción inversa del VIH. Se ha demostrado que la lamivudina actúa aditiva o sinérgicamente con otros agentes anti-VIH, particularmente la zidovudina, inhibiendo la replicación del VIH en cultivos celulares. La lamivudina no interfiere con el metabolismo celular de los desoxinucleótidos, y tiene poco efecto en el contenido de ADN mitocondrial y en las células de mamíferos. En estudios in vitro, la lamivudina ha demostrado poca citotoxicidad para los linfocitos de sangre periférica, para las líneas celulares establecidas de linfocitos y monocitos-macrófagos, y para una variedad de células madre de médula ósea in vitro. Por tanto, la lamivudina tiene un alto índice terapéutico in vitro. La resistencia del VIH-1 a la lamivudina comprende el desarrollo de un cambio en el aminoácido M184V, próximo al sitio activo de la transcriptasa inversa (TI) vírica. Esta variante aparece tanto in vitro como en pacientes infectados con VIH-1 tratados con terapia antirretrovírica que contiene lamivudina. Los mutantes M184V muestran una susceptibilidad significativamente reducida a la lamivudina, y una capacidad replicativa vírica disminuida in vitro. Los estudios in vitro indican que los aislados de virus resistentes a la zidovudina pueden volverse sensibles a la zidovudina cuando adquieren simultáneamente resistencia a la lamivudina. Sin embargo, la importancia clínica de estos hallazgos no se encuentra bien definida. La resistencia cruzada conferida por la TI M184V se encuentra limitada dentro de la clase de agentes antirretrovíricos inhibidores de nucleósidos. La zidovudina y la estavudina mantienen sus actividades antirretrovíricas contra el VIH-1 resistente a la lamivudina. El abacavir mantiene sus actividades antirretrovíricas contra el VIH-1 resistente a la lamivudina que sólo porta la mutación M184V. La TI mutante M184V muestra una disminución 4 veces menor en la susceptibilidad a la didanosina y a la zalcitabina; se desconoce la importancia clínica de estos hallazgos. Las pruebas de susceptibilidad in vitro no se han estandarizado y, asimismo, los resultados pueden variar de acuerdo a los factores metodológicos. En los estudios clínicos, se demostró que la lamivudina, en combinación con zidovudina, reduce la carga vírica del VIH-1 y aumenta el recuento de células CD4. Los datos obtenidos a partir de los criterios de valoración clínica indican que la lamivudina, en combinación con zidovudina sola o en combinación con regímenes de tratamiento que contienen zidovudina, ocasiona una reducción significativa en el riesgo de progresión de la enfermedad y mortalidad. En aislados de VIH obtenidos de pacientes que han recibido terapia con EPIVIR®, se han comunicado casos in vitro de sensibilidad reducida a la lamivudina. Los indicios obtenidos de los estudios clínicos muestran que la lamivudina, adicionada con zidovudina, retarda la aparición de aislados resistentes a la zidovudina en individuos que nunca habían recibido previamente terapia antirretrovírica. La lamivudina se ha utilizado ampliamente como componente de la terapia antirretrovírica de combinación, con otros agentes antirretrovíricos de la misma clase (inhibidores nucleosídicos de la transcriptasa inversa), o de clases diferentes (inhibidores de la proteasa, inhibidores no nucleosídicos de la transcriptasa inversa). Se ha demostrado que la terapia con múltiples fármacos antirretrovíricos, incluyendo lamivudina, es eficaz tanto en pacientes que no han sido tratados previamente con agentes antirretrovíricos, como en pacientes infectados con virus que contienen las mutaciones M184V. Se sigue investigando la relación existente entre la sensibilidad in vitro del VIH a la lamivudina y la respuesta clínica a la terapia. Profilaxis posterior a la exposición (PPE): Las directrices internacionalmente reconocidas (Centro para el Control y Prevención de Enfermedades - Junio de 1998), recomiendan que, en caso de una exposición accidental a sangre infectada por VIH p.ej., al pincharse con la aguja de una jeringa, debe administrarse prontamente una combinación de zidovudina y lamivudina (en un lapso de 1 a 2 horas). En los casos de mayor riesgo de infección, se debe incluir en el régimen algún inhibidor de proteasa. Se recomienda continuar con la profilaxis antirretrovírica durante cuatro semanas. No se han realizado estudios clínicos controlados en la profilaxis posterior a la exposición, por lo que los datos de soporte son limitados. A pesar del rápido tratamiento con agentes antirretrovíricos, aún es posible que ocurra seroconversión. Farmacocinética: Absorción: La lamivudina se absorbe óptimamente del tracto gastrointestinal y, asimismo, la biodisponibilidad de la lamivudina oral en adultos normalmente es de 80 a 85%. Después de la administración oral, el tiempo medio (tmáx) para alcanzar las concentraciones séricas máximas (Cmáx) es de aproximadamente una hora. A niveles terapéuticos de dosificación, es decir, 4 mg/kg/día (en dos dosis, una cada 12 horas), la Cmáx es del orden de 1 a 1.9 microgramos/mL. La coadministración de lamivudina con alimentos ocasiona una demora en la tmáx y una Cmáx menor (disminución de hasta 47%). Sin embargo, la cantidad (con base en el ABC) de lamivudina absorbida permaneció inalterada. No se requieren ajustes en la dosificación cuando la lamivudina se coadministra con los alimentos. No se esperaría que la administración de comprimidos triturados, junto con una pequeña cantidad de líquido o alimento semisólido, tuviese algún impacto en la calidad farmacéutica, por lo cual no se esperaría que alterase el efecto clínico. Esta conclusión se encuentra sustentada en las características fisicoquímicas y farmacocinéticas del ingrediente activo, así como en el comportamiento de la disolución in vitro de las comprimidos de lamivudina en agua, asumiendo que el paciente tritura y transfiere el 100% del comprimido y lo ingiere inmediatamente. Comprimidos: La administración de dos comprimidos de 150 mg es bioequivalente a la administración de una comprimido de 300 mg, con respecto al ABC, Cmáx, y tmáx. Distribución: A partir de los estudios realizados con dosis intravenosas, se determinó que el volumen medio de distribución es de 1.3 l/kg y la vida media terminal promedio de eliminación es de 5 a 7 horas. La lamivudina exhibe una farmacocinética lineal en el intervalo de dosificación terapéutica y, además, muestra un bajo grado de fijación a las albúminas plasmáticas. Los pocos datos disponibles muestran que la lamivudina penetra al sistema nervioso central y llega al líquido cefalorraquídeo (LCR). El cociente medio de concentración de lamivudina en LCR/suero, 2 a 4 horas después de la administración oral, fue de aproximadamente 0.12. Se desconoce el verdadero grado de penetración de la lamivudina o su relación con la eficacia clínica. Metabolismo y Eliminación: La depuración sistémica media de lamivudina es de aproximadamente 0.32 l/h/kg, eliminándose principalmente por la vía renal (más del 70%), a través del sistema de transporte catiónico orgánico y, en menor grado, por metabolismo hepático (menos de un 10%). El compuesto activo, trifosfato de lamivudina intracelular, posee una prolongada vida media terminal en las células (16 a 19 horas), en comparación con la vida media de la lamivudina en el plasma (5 a 7 horas). En 60 voluntarios adultos sanos, se ha demostrado que 300 mg de lamivudina, administrados una vez al día, son farmacocinéticamente equivalentes en estado estacionario a 150 mg de lamivudina administrados dos veces al día, en lo que respecta al ABC24 y a la Cmáx del trifosfato intracelular. La probabilidad de que se presenten interacciones adversas entre la lamivudina y otros medicamentos es baja, ya que tanto la fijación a proteínas plasmáticas como el metabolismo son limitados y, además, casi toda la lamivudina se elimina por la vía renal en forma inalterada. Poblaciones de Pacientes Especiales: Niños: En general, la farmacocinética de la lamivudina en los pacientes pediátricos es similar a la de los adultos. Sin embargo, se redujo la biodisponibilidad absoluta (aproximadamente 55 a 65%) en los pacientes pediátricos menores de 12 años de edad. Además, los valores de depuración sistémica fueron mayores en los pacientes pediátricos más jóvenes y disminuyeron con respecto a la edad, aproximándose a los valores observados en adultos alrededor de los 12 años de edad. Hallazgos recientes indican que el nivel de exposición en niños de 2 a < 6 años de edad podría verse reducido en aproximadamente 30%, en comparación con otros grupos de edad. Actualmente se esperan datos adicionales que respalden esta conclusión. A la fecha, los datos disponibles no sugieren que la lamivudina sea menos eficaz en este grupo de edad. En menos de tres años de edad, los datos disponibles son insuficientes para proponer recomendaciones posológicas específicas. Pacientes de edad avanzada: No se dispone de datos farmacocinéticos para pacientes mayores de 65 años de edad. Insuficiencia renal: Las concentraciones plasmáticas de lamivudina (ABC) aumentan en los pacientes con insuficiencia renal, debido a la disminución en la depuración. Por tanto, se debe reducir la dosificación en aquellos pacientes con una depuración de creatinina inferior a 50 mL/min (ver Dosificación). Insuficiencia hepática: Los datos obtenidos de pacientes con insuficiencia hepática de grado moderado a severo demuestran que la insuficiencia hepática no afecta significativamente la farmacocinética de la lamivudina. Embarazo: La farmacocinética de la lamivudina es similar a la de las mujeres adultas no embarazadas. En los seres humanos, las concentraciones séricas de lamivudina en el bebé al nacer fueron similares a las encontradas en el suero materno y en el cordón umbilical durante el parto, lo cual coincide con la transmisión pasiva de lamivudina a través de la placenta. Datos Preclínicos de Seguridad: Carcinogenicidad, mutagenicidad: La lamivudina no fue mutagénica en pruebas bacterianas pero, al igual que muchos análogos de nucleósido, mostró actividad en un ensayo citogenético in vitro y en el ensayo en linfoma de ratón. La lamivudina no fue genotóxica in vivo, a dosis que produjeron concentraciones plasmáticas aproximadamente 40 a 50 veces superiores a los niveles plasmáticos clínicos anticipados. Como en las pruebas in vivo no se pudo confirmar la actividad mutagénica in vitro de la lamivudina, se concluye que EPIVIR® no debe representar un riesgo genotóxico para los pacientes sometidos al tratamiento. Los estudios de carcinogenicidad a largo plazo, realizados con lamivudina administrada vía oral en ratas y ratones, no mostraron potencial carcinogénico alguno. Toxicología en la reproducción: Los estudios de reproducción realizados en animales no han mostrado indicios de teratogenicidad, ni mostraron efectos en la fertilidad de machos o hembras. La lamivudina produjo pequeños aumentos en la pérdida embrionaria temprana, cuando se administró a conejas preñadas, a niveles de exposición comparables a los alcanzados en los humanos. Sin embargo, no hubo indicios de pérdida embrionaria en ratas expuestas a niveles aproximadamente 35 veces superiores a la exposición clínica (con base en la Cmáx). Toxicología en animales: En los estudios de toxicidad, realizados en animales, la administración de dosis muy elevadas de lamivudina no se asoció con ningún tipo de toxicidad en órganos importantes. Las reducciones en los recuentos de eritrocitos y neutrófilos se identificaron como los efectos con una mayor probabilidad de ser clínicamente importantes.
Indicaciones.
La formulación EPIVIR®, en combinación con otros agentes antirretrovíricos, se indica en el tratamiento de adultos y niños infectados por el VIH.
Dosificación.
La terapia con EPIVIR® debe ser iniciada por un médico con experiencia en el tratamiento de la infección ocasionada por VIH. EPIVIR® puede tomarse con o sin alimentos. Para garantizar la administración de la dosis completa, sería ideal que lo(s) comprimidos(s) fueran(n) deglutidos sin triturarse. Para aquellos pacientes que no puedan deglutir comprimidos, lamivudina se encuentra disponible en solución oral. Alternativamente los comprimidos pueden ser triturados y adicionados a una pequeña cantidad de líquido o alimento semisólido, que deberá consumirse inmediatamente (ver Farmacocinética). Adultos y adolescentes con un peso corporal de cuando menos 30 kg: La dosis recomendada de EPIVIR® consiste en 300 mg al día. Ésta puede administrarse como 150 mg (15 mL de solución oral, 1 comprimido de 150 mg), dos veces al día, o 300 mg (30 mL de solución oral, 2 comprimidos de 150 mg o 1 comprimido de 300 mg) una vez al día. Niños mayores de tres meses de edad y con un peso corporal menor de 30 kg. Solución oral: La dosis recomendada consiste en 4 mg/kg, administrados dos veces al día, hasta un máximo de 300 mg al día. Comprimidos: Niños con un peso corporal entre 21 kg y 30 kg: La dosis oral recomendada de EPIVIR® (150 mg) consiste en medio comprimido tomado en la mañana y un comprimido completo tomado en la noche. Niños con un peso corporal de 14 kg a 21 kg: La dosis oral recomendada de EPIVIR® (150 mg) consiste en medio comprimido ranurado tomado dos veces al día. Niños menores de tres meses de edad: Los pocos datos disponibles resultan insuficientes para establecer recomendaciones específicas de dosificación (ver Farmacocinética). Pacientes de edad avanzada: No se dispone de datos específicos, sin embargo, se recomienda tener un cuidado especial en este grupo de edad debido a los cambios asociados con la edad, como la disminución en la función renal y la alteración de los parámetros hematológicos. Insuficiencia renal: Las concentraciones plasmáticas de lamivudina (ABC) aumentan en los pacientes que exhiben insuficiencia renal de grado moderado a severo, debido a la disminución en la depuración (ver Farmacocinética). Por tanto, la dosis debe reducirse en aquellos pacientes con una depuración de creatinina inferior a 50 mL/min, de acuerdo con la siguiente tabla. Los mismos porcentajes de reducción en la dosificación aplican para los pacientes pediátricos con insuficiencia renal. Cuando se requiere la administración de dosis inferiores a 150 mg, se recomienda emplear la formulación EPIVIR® en solución oral.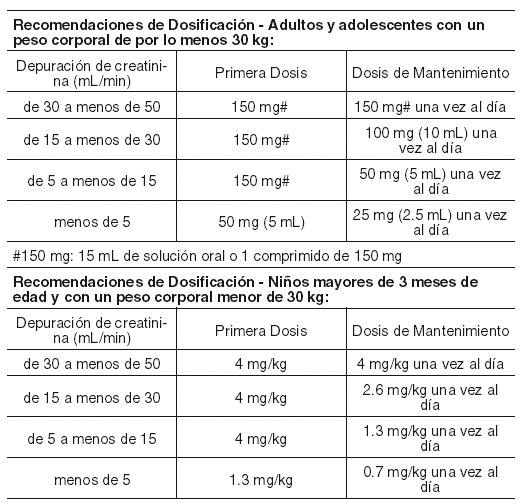
Insuficiencia hepática: No se requiere realizar ajustes en la dosificación de los pacientes con insuficiencia hepática de grado moderado o severo, a menos que el paciente también exhiba insuficiencia renal (ver Farmacocinética).
Contraindicaciones.
El uso de EPIVIR® se contraindica en los pacientes con hipersensibilidad conocida a la lamivudina o a cualquier ingrediente de la preparación.
Reacciones adversas.
Los eventos que se listan a continuación se han comunicado durante el tratamiento de la enfermedad ocasionada por el VIH, con EPIVIR® administrado solo y en combinación con otros agentes antirretrovíricos. En muchos casos, no es claro si esos eventos se relacionan con los medicamentos o si son el resultado del proceso patológico subyacente. Se ha empleado la siguiente convención para la clasificación de los efectos adversos: muy común ( > 1/10), común ( > 1/100, < 1/10), no común ( > 1/1000, < 1/100), raro ( > 1/10000, < 1/1000) muy raro ( < 1/10000). Trastornos sanguíneos y del sistema linfático: No comunes: Neutropenia, anemia, trombocitopenia. Muy raro: Aplasia eritrocítica pura. Trastornos metabólicos y nutricionales: Común: Hiperlactatemia. Raro: Acidosis láctica (ver Advertencias y Precauciones). Redistribución/acumulación de grasa corporal (ver Advertencias y Precauciones). La incidencia de ocurrencia de este evento depende de diversos factores, incluyendo la combinación particular de fármacos antirretrovíricos. Trastornos del sistema nervioso: Común: Cefalea, Insomnio. Muy raros: Parestesia. Se han comunicado casos de neuropatía periférica, aunque no se sabe con precisión si existe alguna relación causal con el tratamiento. Trastornos respiratorios: Común: Tos, síntomas nasales. Trastornos gastrointestinales: Comunes: Náuseas, vómito, dolor en el abdomen superior, diarrea. Raros: Pancreatitis, aunque no se sabe con precisión si existe alguna relación causal con el tratamiento. Aumentos en los niveles séricos de amilasa. Trastornos hepatobiliares: No comunes: Elevaciones transitorias en los niveles de enzimas hepáticas (ASAT, ALAT). Trastornos de la piel y del tejido subcutáneo: Comunes: Exantema, alopecia. Trastornos musculoesqueléticos y del tejido conjuntivo: Comunes: Artralgia, trastornos musculares. Raro: Rabdomiólisis. Trastornos generales y en el sitio de administración: Comunes: Fatiga, malestar general, fiebre.
Advertencias.
No se recomienda el uso de la formulación EPIVIR® como monoterapia. Se debe advertir a los pacientes que aún no se demuestra que la terapia antirretrovírica actual, incluyendo EPIVIR®, evite el riesgo de transmitir el VIH a otras personas a través del contacto sexual o por contaminación sanguínea. Por tanto, se deben seguir tomando las precauciones adecuadas. Existe la posibilidad de que los pacientes tratados con EPIVIR®, o con cualquier otra terapia antirretrovírica, sigan desarrollando infecciones oportunistas y otras complicaciones de la infección por VIH, por lo que deben permanecer bajo estrecha observación clínica por médicos con experiencia en el tratamiento de pacientes con enfermedades asociadas con el VIH. Insuficiencia renal: Las concentraciones plasmáticas de lamivudina (ABC) aumentan en los pacientes con insuficiencia renal de grado moderado a severo, debido a la disminución en su depuración. Por tanto, debe realizarse un ajuste en la dosificación (ver Dosis y Administración). Pancreatitis: Se ha observado pancreatitis en algunos pacientes que reciben EPIVIR®. Sin embargo, no es claro si este trastorno se debió al tratamiento medicamentoso o a la enfermedad por VIH ya existente. Se debe considerar la posibilidad de ocurrencia de pancreatitis cada vez que un paciente desarrolle dolor abdominal, náuseas, vómito o se le detecten marcadores bioquímicos elevados. Se debe suspender la administración de EPIVIR® hasta descartar un diagnóstico de pancreatitis. Acidosis láctica/hepatomegalia severa con esteatosis: Se han comunicado casos de acidosis láctica y hepatomegalia severa con esteatosis, con inclusión de casos mortales, al usar análogos de nucleósido antirretrovíricos, ya sea solos o en combinación, incluyendo lamivudina. La mayoría de estos casos ha tenido lugar en mujeres. Las manifestaciones clínicas que pueden indicar el desarrollo de acidosis láctica incluyen debilidad generalizada, anorexia y pérdida de peso súbita e inexplicable, así como síntomas gastrointestinales y síntomas respiratorios (disnea y taquipnea). Se debe tener precaución al administrar EPIVIR® a cualquier paciente, particularmente a los que se sabe exhiben factores de riesgo de padecer enfermedades hepáticas. Se debe suspender el tratamiento con EPIVIR® en cualquier paciente que desarrolle hallazgos clínicos o de laboratorio que sugieran la presencia de acidosis láctica o hepatotoxicidad (que pueden incluir hepatomegalia y esteatosis, aún en ausencia de elevaciones muy notables en los niveles de aminotransferasas). Los pacientes que presentan coinfección del VIH con hepatitis C y están siendo tratados con interferón alfa y ribavirina, presentan un riesgo aumentado de reacciones adversas severas a nivel hepático, por lo que deben ser supervisados permanentemente. Redistribución de la grasa: En algunos pacientes que reciben terapia antirretrovírica de combinación, se ha observado una redistribución / acumulación de grasa corporal, con inclusión de obesidad central, aumento de la grasa dorsocervical (joroba de búfalo), disminución de la grasa periférica, disminución de la grasa facial, crecimiento mamario, niveles elevados de lípidos séricos y glucosa sanguínea, ya sea por separado o conjuntamente (ver Efectos Adversos). Aunque se ha asociado a todos los miembros de las clases de medicamentos PI (inhibidores de la proteasa) y NRTI (inhibidores nucleosídicos