EPOKINE
PMG PHARMA
Eritropoyético.
Descripción.
Es una solución estéril en frascos de vidrio transparente (vial o jeringa prellenada).
Composición.
Ingrediente activo: eritropoyetina recombinante humana 1.000/2.000/3.000/4.000 y 10.000 unidades. Estabilizador: albúmina sérica humana 2,5mg/ml.
Propiedades.
Es una eritropoyetina recombinante humana tipo alfa, la cual es producida por Cheil Jedang Corporation de Corea, es una hormona glicoproteica que estimula la división y diferenciación de los progenitores eritraideos en la médula ósea. Tiene los mismos efectos inmunológicos y biológicos, así como eritropoyetina endógena, contiene la secuencia del ácido amino de la eritropoyetina aislada neutral.
Indicaciones.
1. En tratamientos de anemia asociada con la falla renal crónica, incluyendo pacientes con diálisis. Está indicado para elevar o mantener los niveles de sangre en las células o en decrecimiento cuando se necesitan transfusiones. 2. Tratamiento en pacientes con cáncer con anemia provocada por quimioterapia. 3. Está indicada para elevar los niveles de sangre en las células para donadores de sangre. 4. También está indicada para prevenir la reducción de hemoglobina para pacientes programados a cirugías mayores, quienes no han participado en un programa de donación de sangre.
Dosificación.
Pacientes con falla renal crónica (FRC): la concentración deseada de hemoglobina debe ser de 10/12/dl en adultos y de 9,6-11g/dl en niños: administrar subcutáneos o intravenoso; dosis media subcutánea 20-30% menor que la dosis intravenosa. En la fase de corrección, incrementar si la hemoglobina no se incrementa a menos de 1g/dl/mes. Un incremento clínicamente significativo no se observa en menos de 2 semanas y puede requerir hasta 6-10 semanas en algunos pacientes. Una vez obtenido el objetivo en la concentración de hemoglobina, disminuir en 25UI/kg/dosis para evitar sobrepasar, el rango esperado. Además, si la concentración de hemoglobina excede en 12g/dl, la terapia debería ser suspendida. Se puede reducir la dosis omitiendo una de las dosis semanales o disminuyendo la cantidad de cada dosis. Pacientes adultos con hemodiálisis: corrección: 50UI/kg tres veces por semana a intervalos de mínimo 4 semanas hasta que la concentración deseada de hemoglobina se alcance. Mantenimiento: entre 30 y 100UI/kg 3 veces por semana. Pacientes con diálisis peritoneal: correctiva: 50UI/kg 2 veces por semana vía subcutánea o intravenosa; incrementos de 25UI/kg 3 veces por semana a intervalos de por lo menos 4 semanas hasta respuesta. Mantenimiento: entre 17 y 33UI/kg 3 veces por semana. Pacientes pediátricos bajo hemodiálisis: corrección: 50UI/kg 3 veces por semana vía intravenosa; incremento de 25UI/kg 3 veces por semana a intervalos de 4 semanas hasta respuesta. Mantenimiento: niños menores de 30kg de peso requieren mayores dosis de mantenimiento que los niños de más de 30kg de peso y que los adultos (75 a 100UI/kg dada por 3 semanas). Pacientes con cáncer: la concentración deseada de hemoglobina debe ser de aprox. 12g/dl. Dosis inicial para prevención o tratamiento de la anemia, 150UI/kg 3 veces por semana vía subcutánea. Si después de 4 semanas de tratamiento, el aumento de la hemoglobina es < 1g/dl, incrementar la dosis hasta 300UI/kg durante 4 semanas. Si después de 4 semanas de terapia con 300UI/kg, la hemoglobina se ha incrementado < 1g/dl, la respuesta es poco probable y el tratamiento debe discontinuarse. Si la hemoglobina aumenta en más de 2g/dl por mes, reducir la dosis en cerca del 25%. Si la hemoglobina se excede de 14g/dl discontinuar la terapia. Fase de corrección: 100UI/kg por tres veces por semana vía subcutánea o intravenosa durante 8 semanas. Fase de mantenimiento: después que la respuesta deseada se obtiene, mantener un hematocrito entre un 30 y un 35% basado en factores tales como, variaciones en la dosis de zidovudina y la presencia de inyecciones intercurrentes o episodios inflamatorios. Si el hematocrito sobrepasa el 40%, discontinuar hasta que el hematocrito disminuya a 36%. Cuando se revisa el tratamiento la dosis debería reducirse en un 25% y luego valorada para mantener el hematocrito deseado. El régimen de dosis recomendado es de 600UI/kg intravenoso 2 veces por semana. Para aquellos pacientes que requieren un grado menor de estimulación eritropoyetica, una dosis de 150-300UI/kg administrada 2 veces por semana, ha demostrado aumentar la predonación autóloga y disminuir la declinación subsecuente en el hematocrito.
Contraindicaciones.
La eritropoyetina está contraindicada en pacientes con: 1) Conocida hipersensibilidad a la droga u otros productos eritropoyéticos. 2) Hipertensión no controlada. 3) Conocida hipersensibilidad a productos derivados de células mamíferas o albúmina humana. 4) Pacientes con antecedentes recientes de infarto al miocardio o accidente vascular cerebral.
Reacciones adversas.
1) Shock: han sido reportados shocks, por lo que deben hacerse observaciones completas. Si los síntomas aparecen, la administración deberá ser descontinuada y deberá darse un tratamiento apropiado. 2) Cardiovascular: hipertensión, trombosis del ducto lagrimal, desviaciones A-V, y taquicardia han sido reportadas raramente. 3) Encefalopatía hipertensiva: tanto encefalopatía hipertensiva (se han demostrado dolores de cabeza, desórdenes en la conciencia y ataques) y hemorragia cerebral han sido reportados ocasionalmente. La droga deberá ser administrada cuidadosamente mediante observaciones del curso de la presión sanguínea y hematocrito durante la terapia. 4) Embolia cerebral: embolias cerebrales han sido reportadas por lo que deberán hacerse observaciones completas, si los síntomas aparecen, deberán discontinuarse la administración y darse un tratamiento adecuado. 5) Piel: comezón, rash de piel, han sido reportados. 6) Hígado: elevación de AST, Alt, LDH, AL-P y bilirrubina pueden ocurrir ocasionalmente. 7) Gastrointestinales: náuseas, vómitos, diarreas y dolores abdominales pueden ocurrir ocasionalmente. 8) Sanguíneos: leucocitosis, eosinofilia, han sido reportados ocasionalmente, en ocasiones pueden ocurrir granulocitopenia en infantes prematuros. Incremento en Bun, de la creatina y el ácido úrico han sido reportados ocasionalmente. 9) Otros: hemorragias cerebrales en los ojos esplenomegalia, hemorragia nasal, edema, dolor de cabeza, mareos, fiebre, fiebres moderadas, fatiga, artralgia, mialgia, tremor y edema pueden ocasionalmente ser asociados con terapia de eritropoyetina. 10) Estudios analizados de datos indican que la eritropoyetina recombinante humana es generalmente bien tolerada. Las reacciones adversas reportadas son frecuentemente secuelas de pacientes enfermos y no son necesariamente atribuibles a terapias con eritropoyetina recombinante humana. Pacientes con falla renal crónica: en estudios de más de 300 pacientes con placebo controlado y con falla renal, las reacciones reportadas en más del 5% de pacientes tratados con eritropoyetina recombinante humana durante la fase, arrojaron los siguientes resultados.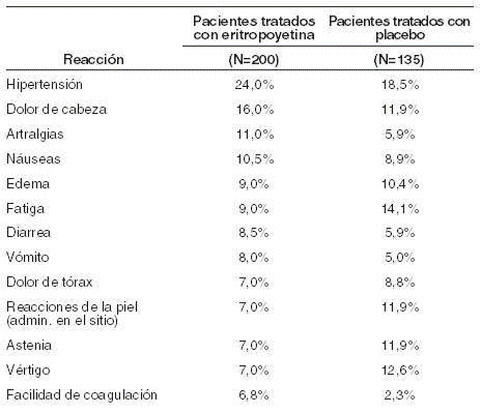
Reacciones adversas significativas de importancia en pacientes con falla renal tratados con: doble-ciego y placebo, ensayos controlados han ocurrido en los siguientes porcentajes durante el estudio en pacientes en la fase: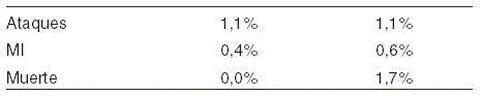
En estudios para pacientes con diálisis aplicando eritropoyetina (N=567), la incidencia de reacciones adversas más frecuentemente reportadas fueron: hipertensión (0,75%), dolor de cabeza (0,40%), taquicardia (0,31%), náuseas y vómitos (0,26%), acceso vascular coagulado (0,25%), respiración disminuida (0,14%), hiperkalemia (0,11%) y diarrea (0,11%). Otras reacciones ocurren en un porcentaje de menos de 0,10% de las reacciones por pacientes durante un año. También se han reportado reacciones ocurridas dentro de varias horas después de la administración con eritropoyetina, esta fue generalmente bien tolerada, independiente de la vía de administración. 2. Pacientes con cáncer en quimioterapia: en estudios ralizados en 131 pacientes con cáncer, tratados con eritropoyetina y placebo, durante tres meses, las reacciones adversas con una incidencia mayor de 10% en cada paciente tratado con eritropoyetina o tratados con placebo, fueron las siguientes.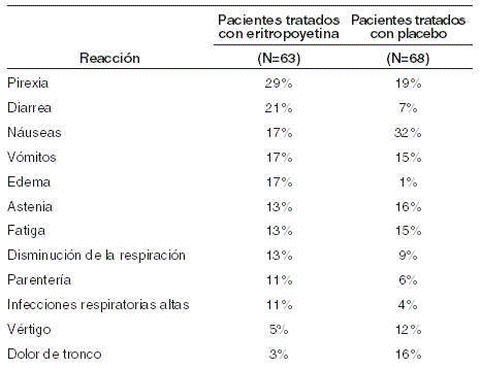
Aunque algunas diferencias satisfactorias significativas fueron notadas entre pacientes tratados con eritropoyetina por ser consistente entre el proceso de enfermedades de cáncer avanzado. Durante la terapia con doble-ciego y subsecuente anteriormente detalladas en las cuales 72 pacientes (N=72) fueron tratados por más de 32 semanas con dosis altas de 927 Unid./kg. Las reacciones adversas perfiladas de eritropoyetina fueron consistentes con la progresión de cáncer avanzado. Basado sobre datos comparables el porcentaje de pacientes sobrevivientes tratados con eritropoyetina y placebo los pacientes que murieron fue debido a los que discontinuaron la terapia, progresión en enfermedades o reacciones adversas (22% y 13% respectivamente: p=0,25), los pacientes tratados con placebo, demostraron ser similares. Datos disponibles desde modelos de tumor en animales y medidas de proliferación de tumores sólidos en células desde espécimen en biopsias clínicas en respuesta a eritropoyetina indican que la eritropoyetina no potencia el crecimiento de tumores. No obstante, como crecimiento de factor, la posibilidad que eritropoyetina pueda potenciar el crecimiento de tumores mieloideos, no pueden ser excluidas. Un estudio controlado de la fase IV corrientemente va sobre la marcha para ampliar la evaluación de estos tejidos. Los principales conteos periféricos de células blancas sanguíneas no fueron cambiados luego de una terapia con eritropoyetina, comparado con los correspondientes valores en el grupo tratado con placebo.
Precauciones.
1) La eritropoyetina deberá ser administrada con precaución en los siguientes pacientes: 1) Pacientes con hipertensión (la presión sanguínea puede subirse o puede ocurrir una encefalopatía hipertensiva durante la terapia con eritropoyetina). 2) Pacientes con historia conocida de hipersensibilidad a la droga. 3) Pacientes con historia conocida de reacciones alérgicas a la droga. 4) Pacientes con infarto al miocardio, infarto pulmonar y embolia cerebral. 5) Pacientes con sangrado cerebral o infantes prematuros con sangrado cerebral.
Advertencias.
1) El tratamiento con eritropoyetina deberá ser limitado en pacientes anémicos con falla renal (FRC) y que tengan menos de 10g/dl de hemoglobina (30% de hematocrito) o en pacientes con cáncer que tengan niveles séricos de eritropoyetina menos de 200mU/ml. 2) La eritropoyetina no podrá ser usada en pacientes con anemia por pérdida de sangre, hematocitopenia e intoxicación con aluminio. 3) Pacientes con historia de shock, deberán hacerse un monitoreo especial para la prevención de shock u otras respuestas. Pequeñas dosis son permitidas para determinar la cantidad o administrar eritropoyetina antes de iniciar la terapia o reasunción después de retenidas. 4) Durante la terapia con eritropoyetina, deberán ser observadas periódicamente las concentraciones de hemoglobina o hematocrito (desde una terapia semanal a dos semanas como terapia de mantenimiento). Se deben tomar precauciones especiales para evitar resultados no excesivos de eritropoyetina (más de 12g/dl de hemoglobina o 36% de hematocrito). En casos de eritropoyesis excesiva, deberá retenerse la droga o en su defecto, administrarse mediante tratamiento adecuado. 5) Hipertensión y encefalopatía hipertensiva han sido reportados en pacientes tratados con eritropoyetina, asociados con un incremento significativo de hematocrito. Los incrementos en hematocrito pueden ocurrir en caso de descontinuación de la terapia. La presión sanguínea en pacientes tratados con eritropoyetina deberán ser cuidadosamente monitoreada, particularmente en pacientes con un historial fundamental de enfermedades de hipertensión o cardiovasculares. La dosis deberá ser ajustada en pacientes con una rápida proporción elevada de hematocrito (mayor de 4% en un período de dos semanas) porque potencialmente el riesgo se incrementa. 6) Han ocurrido ataques en pacientes con insuficiencia renal en pruebas clínicas con eritropoyetina. En pacientes con diálisis, han habido altas incidencias de ataques durante los primeros 90 días de terapia (ocurriendo en aproximadamente 2,5% de pacientes) comparado tiempo después. También han ocurrido ataques en pacientes con cáncer en quimioterapia. En pruebas controladas con placebo doble-ciego, 3,2% (N=2/63) de pacientes tratados con eritropoyetina y 2,9% (N=2/68) de pacientes tratados con placebo, han ocurrido ataques. En un 1,65% (N=1/63) de los pacientes tratados con eritropoyetina ocurrieron ataques en el contexto de un incremento significativo en presión sanguínea y valores bajos de hematocrito. Sin embargo, en ambos pacientes tratados con eritropoyetina también ha habido patologías fundamentales las cuales han sido relacionadas a la actividad de ataques. Dado el potencial incremento en el riesgo de ataques durante la terapia, deberán ser monitoreados exactamente la presión sanguínea y premonitoreada la presencia de síntomas neurológicos. 7) Pueden ocurrir eventos trombolíticos tales como infarto en el miocardio, embolia pulmonar, accidentes cerebro vasculares o ataques de isquemia. Los pacientes con enfermedades vasculares deben ser cuidadosamente monitoreados. 8) Puede ocurrir hiperkalemia por lo que es importante el cumplimiento con las dietas medicinales y reforzar las prescripciones administradas. 9) Monitorear cuidadosamente la circulación sanguínea y equipos de diálisis porque pueden existir residuos sanguíneos. 10) En casos de deficiencia de hierro, debe hacerse un suplemento de hierro como soporte en la eritropoyesis. 11) La eritropoyetina es un factor de crecimiento que primariamente estimula la producción de células rojas, sin embargo, existe la posibilidad de que la eritropoyetina pueda actuar como un factor de crecimiento para algún tipo de tumores, particularmente los tumores mieloideos malignos. Usos en mujeres embarazadas: la seguridad de eritropoyetina en mujeres embarazadas no ha sido establecida. La eritropoyetina puede ser usada durante el embarazo, solo si el potencial beneficia o justifica el potencial de riesgo. Uso pediátrico: la seguridad de la eritropoyetina en niños no ha sido establecida. Usos en geriatría: si la eritropoyetina es administrada en pacientes geriátricos, la frecuencia de la dosis deberá ser controlada sobre las bases de la observación de la presión sanguínea y la concentración de la hemoglobina o hematocrito. Precauciones y administración: 1) no se diluya o administre en conjunto con otras drogas en soluciones. 2) Si administra eritropoyetina en pacientes después de la diálisis o con diálisis o con síntomas de gripe, hacerlo inyectándolo lentamente durante cinco minutos. 3) La eritropoyetina no deberá administrarse por infusión. Si se usa la vía intravenosa, deberá ser administrada lentamente.
Conservación.
Almacenamiento: guardar a una temperatura ambiente de 2° a 8°C y proteger de la luz. Vida útil: período de vida 18 meses.
Sobredosificación.
La respuesta de la dosis de eritropoyetina depende sobretodo de las condiciones del paciente. En caso de sobredosis, puede ocurrir hipertensión, si la policitemia es de importancia puede indicarse flebotomía para el decrecimiento de los hematocrito.
Presentación.
La eritropoyetina está disponible en cartones de 6 jeringas prellenadas de 1.000UI de 0,5ml o en frasco ampolla de 2.000UI de 1ml, o 4.000UI de 1ml en cartones de 10 frascos ampollas.