FESATEROX
EXELTIS
Trat. síndrome de vejiga hiperactiva.
Composición.
FESATEROX comprimidos recubiertos de liberación prolongada 4 mg. Cada comprimido recubierto de liberación prolongada contiene 4mg de fumarato de fesoterodina, correspondiente a 3,1 mg de fesoterodina. FESATEROX comprimidos recubiertos de liberación prolongada 8 mg. Cada comprimido recubierto de liberación prolongada contiene 8 mg de fumarato de fesoterodina, correspondiente a 6,2 mg de fesoterodina. Excipientes con efecto conocido: FESATEROX comprimidos recubiertos de liberación prolongada 4 mg. Cada comprimido recubierto de liberación prolongada contiene 72 mg de fructosa y 58 mg de lactosa (como monohidrato). FESATEROX comprimidos recubiertos de liberación prolongada 8 mg. Cada comprimido recubierto de liberación prolongada contiene 72 mg de fructosa y 55 mg de lactosa (como monohidrato). Forma farmacéutica: Comprimidos recubiertos de liberación prolongada. FESATEROX comprimidos recubiertos de liberación prolongada 4 mg: Azul, elíptico, comprimido recubierto biconvexo con aproximadamente 6 mm de diámetro y grabado con "F4" por un lado. FESATEROX comprimidos recubiertos de liberación prolongada 8 mg: Azul oscuro, elíptico, comprimido recubierto biconvexo con aproximadamente 6 mm de diámetro y grabado con "F8" por un lado.
Farmacología.
Propiedades farmacodinámicas: Grupo farmacoterapéutico: Urológicos, drogas para la incontinencia y frecuencia urinaria., código ATC: G04BD11. Mecanismo de acción: La fesoterodina es un antagonista competitivo y específico de los receptores muscarínicos. Se hidroliza rápida y extensamente por esterasas plasmáticas no específicas al derivado 5-hidroximetilo, su principal metabolito activo, que es el principal principio farmacológico activo de la fesoterodina. Eficacia clínica y seguridad: La eficacia de dosis fijas de fesoterodina de 4 mg y 8 mg se evaluó en dos ensayos de Fase 3, aleatorizados, doble ciego, controlados con placebo, de 12 semanas. Se incluyeron pacientes mujeres (79%) y hombres (21%) con una edad media de 58 años (rango 19-91 años). Un total de 33% de los pacientes tenían ≥65 años de edad y 11% tenían ≥75 años de edad. Los pacientes tratados con fesoterodina tuvieron reducciones medias estadísticamente significativas en el número de micciones por 24 horas y en el número de episodios de incontinencia de urgencia por 24 horas al final del tratamiento, en comparación con el placebo. Del mismo modo, la tasa de respuesta (% de pacientes que informaron que su condición había "mejorado mucho" o "mejorado" utilizando una escala de beneficios del tratamiento de 4 puntos) fue significativamente mayor con fesoterodina en comparación con placebo. Además, la fesoterodina mejoró el cambio medio en el volumen evacuado por micción y el cambio medio en el número de días continencia por semana (consulte la Tabla 1). Tabla 1: Cambios medios desde el inicio hasta el final del tratamiento para los criterios de valoración primarios y secundarios seleccionados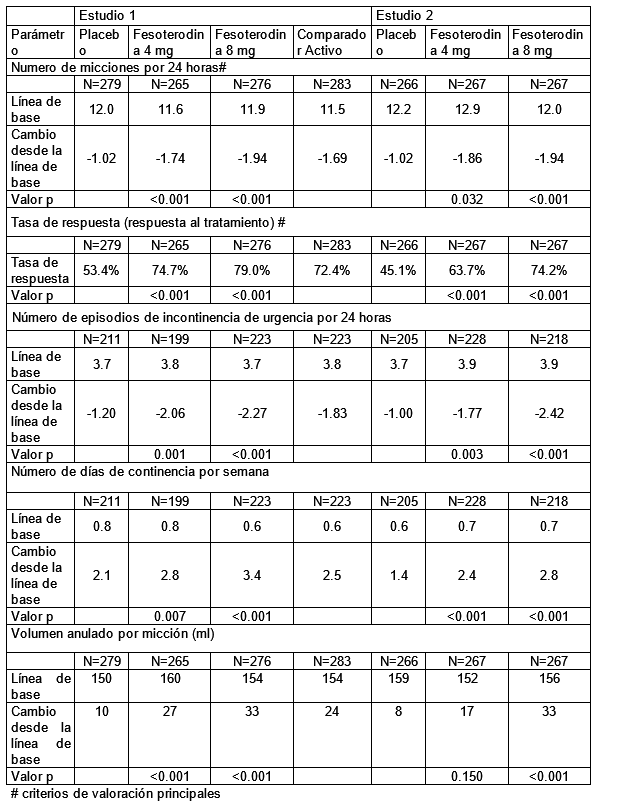
Electrofisiología cardíaca: El efecto de fesoterodina 4 mg y 28 mg sobre el intervalo QT se evaluó minuciosamente en un ensayo de grupos paralelos doble ciego, aleatorizado, placebo - y positivo - controlado (moxifloxacino 400 mg), con tratamiento una vez al día durante un período de 3 días en 261 sujetos masculinos y femeninos de 45 a 65 años. El cambio desde el inicio en QTc basado en el método de corrección de Fridericia no mostró ninguna diferencia entre el grupo de tratamiento activo y el de placebo. Propiedades Farmacocinéticas: Absorción: Después de la administración oral, debido a la rápida y extensa hidrólisis por esterasas plasmáticas no específicas, no se detectó fesoterodina en plasma. La biodisponibilidad del metabolito activo es del 52%. Después de la administración oral de dosis únicas o múltiples de fesoterodina en dosis de 4 mg a 28 mg, las concentraciones plasmáticas del metabolito activo son proporcionales a la dosis. Los niveles plasmáticos máximos se alcanzan después de aproximadamente 5 horas. Los niveles plasmáticos terapéuticos se alcanzan después de la primera administración de fesoterodina. No se produce acumulación después de la administración de dosis múltiples. Distribución: La unión a proteínas plasmáticas del metabolito activo es baja y aproximadamente el 50 % se une a la albúmina y la alfa-1-glucoproteína ácida. El volumen medio de distribución en estado estacionario después de la infusión intravenosa del metabolito activo es de 169 l. Biotransformación: Después de la administración oral, la fesoterodina se hidroliza rápida y ampliamente a su metabolito activo. El metabolito activo se metaboliza aún más en el hígado a su metabolito carboxi, carboxi-N-desisopropilo y N-desisopropilo con participación de CYP2D6 y CYP3A4. Ninguno de estos metabolitos contribuye significativamente a la actividad antimuscarínica de la fesoterodina. La Cmax y el AUC medias del metabolito activo son 1,7 y 2 veces mayores, respectivamente, en los metabolizadores lentos de CYP2D6 en comparación con los metabolizadores rápidos. Eliminación: El metabolismo hepático y la excreción renal contribuyen significativamente a la eliminación del metabolito activo. Después de la administración oral de fesoterodina, aproximadamente el 70 % de la dosis administrada se recuperó en la orina como metabolito activo (16 %), metabolito carboxi (34 %), metabolito carboxi-N-desisopropilo (18 %) o metabolito N-desisopropilo (1 %) y una cantidad menor (7 %) se recuperó en las heces. La vida media terminal del metabolito activo después de la administración oral es de aproximadamente 7 horas y está limitada por la tasa de absorción. Edad y género:_No se recomienda ajuste de dosis en estas subpoblaciones. La farmacocinética de fesoterodina no está significativamente influenciada por la edad y el sexo. Población pediátrica: No se ha evaluado la farmacocinética de fesoterodina en pacientes pediátricos. Insuficiencia renal: En pacientes con insuficiencia renal leve o moderada (TFG 30 - 80 ml/min), la Cmax y el AUC del metabolito activo aumentaron hasta 1,5 y 1,8 veces, respectivamente, en comparación con sujetos sanos. En pacientes con insuficiencia renal grave (TFG < 30 ml/min), la Cmax y el AUC aumentan 2,0 y 2,3 veces, respectivamente. Insuficiencia hepática: En pacientes con insuficiencia hepática moderada (Child Pugh B), la Cmax y el AUC del metabolito activo aumentaron 1,4 y 2,1 veces, respectivamente, en comparación con sujetos sanos. No se ha estudiado la farmacocinética de fesoterodina en pacientes con insuficiencia hepática grave. 5.3 Datos preclínicos de seguridad: En estudios no clínicos de farmacología de seguridad, toxicidad general, genotoxicidad y carcinogenicidad no se han observado efectos clínicamente relevantes, excepto los relacionados con el efecto farmacológico del principio activo. Los estudios de reproducción han mostrado una embriotoxicidad menor a dosis cercanas a las tóxicas para la madre (aumento del número de reabsorciones, pérdidas pre y post implantación). Se ha demostrado que las concentraciones supraterapéuticas del metabolito activo de la fesoterodina inhiben la corriente K+ en los canales Herg-huma ether-a-go-go-realted gene y prolongan la duración del potencial de acción (70% y 90% de repolarización) en las fibras caninas aisladas de Purkinje. Sin embargo, en perros conscientes, el metabolito activo no tuvo ningún efecto sobre el intervalo QT y el intervalo QTc en exposiciones plasmáticas, al menos, 33 veces superiores a la concentración plasmática libre máxima media en humanos que son metabolizadores extensos; y 21 veces superior a la medida en sujetos que son metabolizadores pobres de CYP2D6 tras la administración de 8 mg de fesoterodina una vez al día. En un estudio sobre la fertilidad y el desarrollo embrionario temprano en ratones, la fesoterodina no tuvo ningún efecto sobre la función reproductora o la fertilidad de los machos en dosis de hasta 45 mg/kg/día. Con 45 mg/kg/día, se observó un menor número de cuerpos lúteos, lugares de implantación y fetos viables en ratones hembra a los que se administró fesoterodina durante 2 semanas antes del apareamiento y hasta el día 7 de la gestación. El nivel sin efecto observado (NOEL) materno y el NOEL para los efectos sobre la reproducción y el desarrollo embrionario temprano fueron ambos de 15 mg/kg/día. Basándose en el AUC, la exposición sistémica fue de 0,6 a 1,5 veces mayor en los ratones que en los humanos en el MRHD, mientras que, basándose en las concentraciones plasmáticas máximas, la exposición en los ratones fue de 5 a 9 veces mayor.
Indicaciones.
FESATEROX está indicada en adultos para el tratamiento de los síntomas (aumento de la frecuencia urinaria y/o de la urgencia urinaria y/o de la incontinencia de urgencia) que pueden producirse con el síndrome de vejiga hiperactiva.
Dosificación.
Posología: Adultos (incluyendo personas de edad avanzada): La dosis inicial recomendada es de 4 mg una vez al día. Según la respuesta individual, la dosis puede aumentarse a 8 mg una vez al día. La dosis diaria máxima es de 8 mg. El efecto completo del tratamiento se observó entre 2 y 8 semanas. Por lo tanto, se recomienda volver a evaluar la eficacia para el paciente individual después de 8 semanas de tratamiento. En sujetos con función renal y hepática normal que reciben administración concomitante de inhibidores potentes de CYP3A4, la dosis diaria máxima de FESATEROX debe ser de 4 mg, una vez al día. Población especial: Insuficiencia renal y hepática: La siguiente tabla proporciona las recomendaciones de dosificación diaria para sujetos con insuficiencia renal o hepática en ausencia y presencia de inhibidores moderados y potentes de CYP3A4.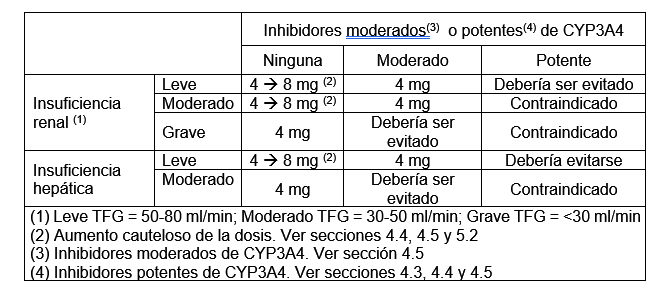
FESATEROX está contraindicada en personas con insuficiencia hepática grave. Población pediátrica: No se ha establecido aún la seguridad y eficacia de FESATEROX en niños menores de 18 años. No hay datos disponibles. Método de administración: Los comprimidos deben tomarse una vez al día con líquido y tragarse; no deben dividirse, triturarse, disolverse, chuparse ni masticarse. FESATEROX puede administrarse con o sin alimentos.
Contraindicaciones.
Hipersensibilidad al principio activo o a alguno de los excipientes. Retención urinaria. Retención gástrica. Glaucoma de ángulo estrecho no controlado. Miastenia gravis. Insuficiencia hepática grave (Child PughC). Uso concomitante de inhibidores potentes de CYP3A4 en personas con insuficiencia hepática o renal, de moderada a grave. Colitis ulcerosa grave. Megacolon tóxico.
Efectos indeseables.
Resumen del perfil de seguridad: La seguridad de fesoterodina se evaluó en ensayos clínicos controlados con placebo en un total de 2859 pacientes con vejiga hiperactiva, de los cuales 780 recibieron placebo. Debido a las propiedades farmacológicas de la fesoterodina, el tratamiento puede causar efectos antimuscarínicos de leves a moderados, como sequedad de boca, sequedad ocular, dispepsia y estreñimiento. La retención urinaria puede ocurrir con poca frecuencia. La sequedad de boca, fue la única reacción adversa muy frecuente, se produjo con una frecuencia del 28,8 % en el grupo de fesoterodina frente al 8,5 % en el grupo de placebo. La mayoría de las reacciones adversas ocurrieron durante el primer mes de tratamiento, con la excepción de los casos clasificados como retención urinaria u orina residual posterior a la micción superior a 200 mL, que podría ocurrir después de un tratamiento a largo plazo y fue más común en hombres que en mujeres. Lista tabulada de reacciones adversas: La siguiente tabla muestra la frecuencia de las reacciones adversas emergentes del tratamiento de los ensayos clínicos controlados con placebo y de la experiencia posterior a la comercialización. Las reacciones adversas se informan en esta tabla con la siguiente convención de frecuencia: muy frecuentes (≥1/10), frecuentes (≥1/100 a < 1/10), poco frecuentes (≥1/1000 a < 1/100), raro (≥1/10000 a < 1/1000). Dentro de cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.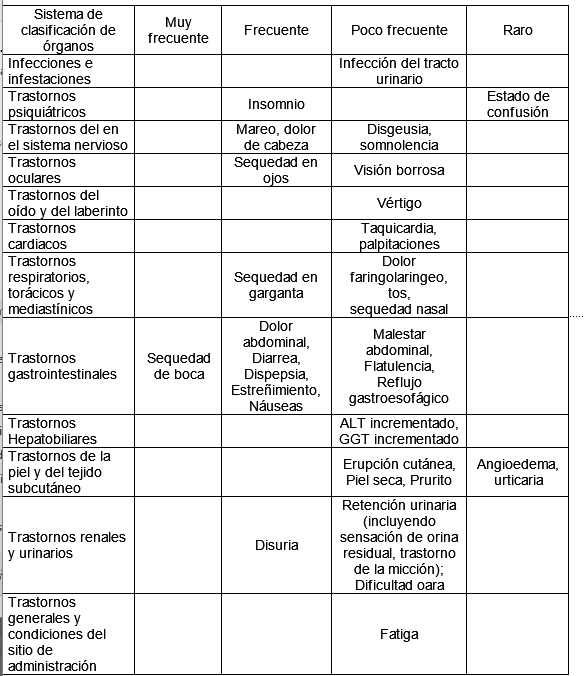
Descripción de reacciones adversas seleccionadas: En los ensayos clínicos de fesoterodina, se informaron casos de enzimas hepáticas marcadamente elevadas con una frecuencia de ocurrencia similar a la del grupo placebo. La relación con el tratamiento con fesoterodina no está clara. Se obtuvieron electrocardiogramas de 782 pacientes tratados con 4 mg, 785 tratados con 8 mg y 222 tratados con 12 mg de fesoterodina; además de 780 pacientes con placebo. El intervalo QT corregido por la frecuencia cardíaca en los pacientes tratados con fesoterodina no difirió del observado en los pacientes tratados con placebo. Las tasas de incidencia de QTc ≥500 ms después de la línea de base o aumento de QTc de ≥60 ms es del 1,9 %, 1,3 %, 1,4 % y 1,5 %, para fesoterodina 4 mg, 8 mg, 12 mg y placebo, respectivamente. La relevancia clínica de estos hallazgos dependerá de los factores de riesgo y susceptibilidades individuales del paciente presentes. Se han descrito casos de retención urinaria que requieren de sondaje, generalmente dentro de la primera semana de tratamiento con fesoterodina. Han afectado principalmente a pacientes varones de edad avanzada (≥ 65 años) con antecedentes compatibles con hiperplasia prostática benigna. Notificación de sospechas de reacciones adversas: Es importante informar de las sospechas de reacciones adversas del medicamento después de su autorización. Permite el seguimiento continuado del balance beneficio/riesgo del medicamento. Se solicita a los profesionales sanitarios que notifiquen cualquier sospecha de reacción adversa a través del sistema nacional de notificación.
Advertencias.
FESATEROX debe utilizarse con precaución en pacientes con: Obstrucción significativa del tracto de salida vesical con riesgo de retención urinaria (p. ej. agrandamiento de la próstata clínicamente significativo debido a hiperplasia prostática benigna). Trastornos obstructivos gastrointestinales (por ejemplo, estenosis pilórica). Reflujo gastroesofágico y/o que estén tomando al mismo tiempo medicamentos (como los bisfosfonatos orales) que pueden causar o exacerbar la esofagitis. Disminución de la motilidad gastrointestinal. Neuropatía autonómica. Glaucoma de ángulo estrecho controlado. Se debe tener precaución al prescribir o aumentar la dosis de fesoterodina a pacientes en los que se espera una mayor exposición al metabolito activo: Insuficiencia hepática. Insuficiencia renal. Administración concomitante de inhibidores potentes o moderados de CYP3A4. Administración concomitante de un inhibidor potente de CYP2D6. Incremento de dosis: En pacientes con una combinación de estos factores, se esperan aumentos de exposición adicionales. Es probable que se produzcan reacciones adversas antimuscarínicas dependientes de la dosis. En poblaciones en las que la dosis puede aumentarse a 8 mg una vez al día, el aumento de la dosis debe ir precedido de una evaluación de la respuesta individual y la tolerabilidad. Las causas orgánicas deben excluirse antes de considerar cualquier tratamiento con antimuscarínicos. Aún no se ha establecido la seguridad y la eficacia en pacientes con una causa neurogénica de hiperactividad del detrusor. Se deben evaluar otras causas de micción frecuente (tratamiento de insuficiencia cardíaca o enfermedad renal) antes del tratamiento con FESATEROX. Si hay infección del tracto urinario, se debe tomar un enfoque médico apropiado o una terapia antibacteriana. Angioedema: Se ha notificado casos de angioedema con el uso de fesoterodina y se ha producido después de la primera dosis en algunos casos. Si se produce angioedema, se debe suspender la fesoterodina y se debe proporcionar de inmediato la terapia adecuada. Inductores potentes de CYP3A4: No se recomienda el uso concomitante de fesoterodina con un inductor potente de CYP3A4 (es decir, carbamazepina, rifampicina, fenobarbital, fenitoína, hierba de San Juan). Prolongación de QT: FESATEROX debe utilizarse con precaución en pacientes con riesgo de prolongación del intervalo QT (por ejemplo: hipopotasemia, bradicardia y administración concomitante de medicamentos que se sabe que prolongan el intervalo QT) y enfermedades cardíacas preexistentes relevantes (por ejemplo: isquemia miocárdica, arritmia, insuficiencia cardíaca congestiva), Esto es especialmente cierto cuando se toman inhibidores potentes de CYP3A4. Lactosa: Los pacientes con problemas hereditarios raros de intolerancia a la galactosa, deficiencia total de lactasa o problemas de absorción de glucosa o galactosa, no deben tomar este medicamento. Fructosa: Este medicamento contiene 72 mg de fructosa por comprimido. Los pacientes con intolerancia hereditaria a la fructosa (IHF) no deben tomar/recibir este medicamento.
Interacciones.
Interacciones farmacológicas: Se debe tener precaución en la administración conjunta de fesoterodina con otros antimuscarínicos y medicamentos con propiedades anticolinérgicas (por ejemplo: amantadina, antidepresivos tricíclicos, ciertos neurolépticos), ya que esto puede provocar efectos secundarios y terapéuticos más pronunciados (por ejemplo: estreñimiento, sequedad de boca, somnolencia, retención urinaria). La fesoterodina puede reducir el efecto de los medicamentos que estimulan la motilidad del tracto gastrointestinal, como la metoclopramida. Interacciones farmacocinéticas: Los datos in vitro demuestran que el metabolito activo de fesoterodina no inhibe CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 o 3A4, ni induce CYP1A2, 2B6, 2C9, 2C19 o 3A4 a concentraciones plasmáticas clínicamente relevantes. Por lo tanto, es poco probable que fesoterodina altere el aclaramiento de medicamentos que son metabolizados por estas enzimas. Inhibidores de CYP3A4: Inhibidores potentes de CYP3A4: Tras la inhibición de CYP3A4 mediante la co-administración de ketoconazol 200 mg dos veces al día, la Cmax y el AUC del metabolito activo de fesoterodina aumentaron 2,0 y 2,3 veces en los metabolizadores rápidos de CYP2D6; y 2,1 y 2,5 veces en los metabolizadores lentos de CYP2D6, respectivamente. Por lo tanto, la dosis máxima de fesoterodina debe restringirse a 4 mg cuando se usa concomitantemente con inhibidores potentes de CYP3A4 (por ejemplo: atazanavir, claritromicina, indinavir, itraconazol, ketoconazol, nefazodona, nelfinavir, ritonavir (y todos los regímenes con inhibidores de proteasa IP potenciados con ritonavir), saquinavir y telitromicina. Inhibidores moderados de CYP3A4: Después del bloqueo de CYP3A4 mediante la coadministración del inhibidor moderado de CYP3A4, fluconazol, 200 mg dos veces al día durante 2 días, la Cmax y el AUC del metabolito activo de fesoterodina aumentaron aproximadamente un 19 % y un 27 %, respectivamente. No se recomiendan ajustes de dosis en presencia de inhibidores moderados de CYP3A4 (por ejemplo: eritromicina, fluconazol, diltiazem, verapamilo y jugo de pomelo). Inhibidores débiles de CYP3A4: No se examinó el efecto de los inhibidores débiles de CYP3A4 (por ejemplo, cimetidina); no se espera que supere el efecto del inhibidor moderado. Inductores de CYP3A4: Después de la inducción de CYP3A4 mediante la coadministración de 600 mg de rifampicina una vez al día, la Cmax y el AUC del metabolito activo de fesoterodina disminuyó aproximadamente un 70% y un 75% respectivamente, después de la administración oral de 8 mg de fesoterodina. La inducción de CYP3A4 puede conducir a niveles plasmáticos subterapéuticos. No se recomienda el uso concomitante con inductores de CYP3A4 (por ejemplo, carbamazepina, rifampicina, fenobarbital, fenitoína, hierba de San Juan). Inhibidores de CYP2D6: La interacción con los inhibidores de CYP2D6 no se probó clínicamente. La Cmax y el AUC medias del metabolito activo son 1,7 y 2 veces mayores (respectivamente) en los metabolizadores lentos de CYP2D6 en comparación con los metabolizadores rápidos. La administración conjunta de un inhibidor potente de CYP2D6 puede resultar en una mayor exposición y eventos adversos. Puede ser necesaria una reducción de la dosis a 4 mg. Anticonceptivos orales: La fesoterodina no afecta la supresión de la ovulación por la anticoncepción hormonal oral. En presencia de fesoterodina no hay cambios en las concentraciones plasmáticas de los anticonceptivos orales combinados que contienen etinilestradiol y levonorgestrel. Warfarina: Un ensayo clínico en voluntarios sanos ha demostrado que la fesoterodina 8 mg una vez al día no tiene un efecto significativo sobre la farmacocinética o la actividad anticoagulante de una dosis única de warfarina. Población pediátrica: Los estudios de interacción solo se han realizado en adultos. Fertilidad, embarazo y lactancia: Embarazo: No hay datos suficientes sobre el uso de fesoterodina en mujeres embarazadas. Los estudios de toxicidad reproductiva con fesoterodina en animales muestran embriotoxicidad menor. En estudios de reproducción en animales, la administración oral de fesoterodina a ratones y conejas preñadas durante la organogénesis resultó con fetotoxicidad en exposiciones maternas, que fueron 6 y 3 veces la dosis máxima recomendada en humanos (MRHD), respectivamente, según el AUC (ver sección 5.3). Se desconoce el riesgo potencial para los humanos. FESATEROX no se recomienda durante el embarazo. Lactancia: Se desconoce si fesoterodina/metabolitos son excretados en la leche humana; por lo tanto, no se recomienda amamantar durante el tratamiento con FESATEROX. Fertilidad: No se han realizado ensayos clínicos para evaluar el efecto de la fesoterodina en la fertilidad humana. Los hallazgos en ratones a exposiciones de aproximadamente 5 a 19 veces las de DMRH muestran un efecto sobre la fertilidad femenina; sin embargo, se desconocen las implicaciones clínicas de estos hallazgos en animales (ver sección 5.3). Las mujeres en edad fértil deben ser conscientes de la falta de datos sobre la fertilidad humana, y FESATEROX solo debe administrarse después de considerar los riesgos y beneficios individuales. Efectos sobre la capacidad para conducir y utilizar máquinas: FESATEROX tiene una influencia menor sobre la capacidad para conducir y utilizar máquinas. Se debe tener precaución al conducir o utilizar máquinas debido a la posible aparición de efectos adversos como visión borrosa, mareos y somnolencia.
Incompatibilidades.
No aplica
Conservación.
No almacenar a más de 25°C. Almacenar en el blister original para protegerlo de la humedad. Vida útil: 2 años
Sobredosificación.
La sobredosis de antimuscarínicos, incluida la fesoterodina, puede provocar efectos anticolinérgicos graves. El tratamiento debe ser sintomático y de soporte. En caso de sobredosis, se recomienda monitorización ECG; deben adoptarse medidas estándar de apoyo para controlar la prolongación del intervalo QT. La fesoterodina se ha administrado de forma segura en ensayos clínicos en dosis de hasta 28 mg/día. En caso de sobredosis de fesoterodina, se debe tratar con lavado gástrico y administrar carbón activado. Trate los síntomas de la siguiente manera: Efectos anticolinérgicos centrales graves (por ejemplo: alucinaciones, exaltación intensa): tratar con fisostigmina. Convulsiones o excitación pronunciada: tratar con benzodiazepinas. Insuficiencia respiratoria: tratar con respiración artificial. Taquicardia: tratar con betabloqueantes. Retención urinaria: tratar con cateterismo..Midriasis: tratar con colirio de pilocarpina y/o colocar al paciente en cuarto oscuro.
Presentación.
FESATEROX comprimidos de recubiertos de liberación prolongada de 4 mg y 8 mg se presentan en blísteres de aluminio en cajas que contienen 10,14, 28, 30, 56, 84, 98 o 100 comprimidos recubiertos de liberación prolongada. No se pueden comercializar todos los tamaños de envase.