GENOTROPIN®
PFIZER
Hormonoterapia.
Composición.
Genotropin® contiene Somatropina humana recombinante 16 UI y 36 UI. Excipientes: Glicina, Manitol, Fosfato de sodio dihidrógeno monohidrato, Fosfato disódico dodecahidrato, c.s. Solvente: M-cresol, Manitol, Agua para inyectables, c.s.
Toxicología.
En estudios relacionados con la toxicidad general, la tolerancia local y la toxicidad de la reproducción no se han observado efectos clínicamente relevantes. Los estudios de genotoxicidad in vitro e in vivo sobre mutaciones genéticas e inducción de aberraciones cromosómicas han sido negativos. Se ha observado un aumento de la fragilidad de los cromosomas en un estudio in vitro sobre linfocitos obtenidos de pacientes después de tratamiento a largo plazo con somatropina y luego de la adición del fármaco radiomimético bleomicina. No está claro el significado clínico de este hallazgo. En otro estudio, no se encontró aumento de las anormalidades cromosómicas en los linfocitos de pacientes que habían recibido terapia con somatropina a largo plazo.
Indicaciones.
Pacientes pediátricos: GENOTROPIN (somatropina [origen ADNr] inyectable) está indicada para el tratamiento de pacientes pediátricos con: Retraso del crecimiento debido a la secreción inadecuada de hormona de crecimiento endógena. Retraso del crecimiento debido al síndrome de Prader-Willi. El diagnóstico del SPW se debe confirmar mediante el análisis genético apropiado. Trastorno del crecimiento (altura actual < -2,5 DE y ajuste de altura respecto los progenitores de < -1 DE) en niños nacidos pequeños para su edad gestacional (PEG) con un peso y/o longitud en el momento de su nacimiento por debajo de -2 DE, que no hayan mostrado una recuperación en el crecimiento (velocidad de crecimiento < 0 DE durante el último año) a los 4 años o posteriormente. Retraso del crecimiento asociado con síndrome de Turner o insuficiencia renal crónica. Talla baja idiopática (TBI) también conocida como talla baja no debida a deficiencia de hormona de crecimiento, que se define por una desviación estándar de la altura < -2,25, y está asociada a tasas de crecimiento que probablemente no permitirán alcanzar la estatura adulta dentro del intervalo normal, en pacientes pediátricos cuyas epífisis no están cerradas y para quienes la evaluación de diagnóstico excluye otras causas asociadas con talla baja que debieran observarse o tratarse por otros medios. Pacientes adultos: GENOTROPIN (somatropina [origen ADNr] inyectable) está indicado en terapia sustitutiva en adultos con un déficit marcado de hormona de crecimiento. Los pacientes con marcado déficit de hormona de crecimiento en la edad adulta están clasificados como pacientes con enfermedad hipotalámico- hipofisiaria con al menos otro déficit conocido hormonal hipofisiario (no siendo el de prolactina). En estos pacientes se deberá realizar un test de estimulación dinámica para diagnosticar o excluir un déficit de hormona de crecimiento. En pacientes con déficit aislado de hormona de crecimiento instaurado en la infancia (sin evidencia de enfermedad hipotalámica- pituitaria o radiación craneal), se recomienda realizar dos test de estimulación dinámica, excepto en aquellos pacientes con concentraciones bajas del factor de crecimiento tipo Insulina I (IGF-I) ( < 2 SDS) en los que deberá realizarse un solo test. El punto de corte del test de estimulación dinámica deberá ser estricto.
Dosificación.
GENOTROPIN no debe inyectarse por vía intravenosa. La inyección deberá administrarse por vía subcutánea y el lugar de inyección deberá variarse para evitar lipoatrofia. La terapia con GENOTROPIN debe estar supervisada por un médico con experiencia en diagnóstico y manejo de pacientes pediátricos con retraso del crecimiento asociado con deficiencia de hormona de crecimiento (DHC), síndrome de Prader-Willi (SPW), síndrome de Turner (ST), que nacieron pequeños para su edad gestacional (PEG) o tienen talla baja idiopática (TBI) y pacientes adultos con DHC con inicio en la infancia o en la edad adulta. Posología en pacientes pediátricos: Información general de posología en pacientes pediátricos El calendario de posología y administración de GENOTROPIN debe individualizarse según la respuesta de crecimiento de cada paciente. La respuesta a la somatropina en pacientes pediátricos tiende a descender con el tiempo. No obstante, en los pacientes pediátricos, el fracaso en incrementar la tasa de crecimiento, especialmente durante el primer año de terapia, indica la necesidad de evaluar de cerca el cumplimiento y de evaluar otras causas de fracaso del crecimiento, como el hipotiroidismo, la subnutrición, la edad ósea avanzada y los anticuerpos a la HC recombinante humana (HCrh). El tratamiento con GENOTROPIN para la talla baja debe interrumpirse cuando se cierran las epífisis (cartílagos de crecimiento). Trastornos del crecimiento debido a secreción insuficiente de hormona de crecimiento en niños: se recomienda una dosis de 0,025-0,035 mg/kg peso corporal/día o 0,7-1,0 mg/m2 de superficie corporal/día. Se han utilizado dosis superiores. Síndrome de Prader-Willi, para mejorar el crecimiento y la composición corporal: Se recomienda una dosis de 0,035 mg/kg de peso corporal al día o 1,0 mg/m2 de superficie corporal día. No se deberá exceder de una dosis de diaria de 2,7 mg. El tratamiento no deberá ser utilizado en niños con una velocidad de crecimiento inferior a 1 cm por año y próximos al cierre de la epífisis. Trastorno de crecimiento asociado a Síndrome de Turner: Se recomienda una dosis de 0,045-0,050 mg/kg de peso corporal día o 1,4 mg/m2 de superficie corporal día. Trastorno del crecimiento debido a Insuficiencia renal crónica: Se recomienda una dosis de 1,4 mg/m2 de superficie corporal día (aproximadamente 0,045- 0,050 mg/kg de peso/día). Pueden ser necesarias dosis más altas si la velocidad de crecimiento es demasiado baja. Una corrección de la dosis puede ser necesaria después de 6 meses de tratamiento. Trastorno del crecimiento en niños nacido pequeños para su edad gestacional (PEG): Se recomienda una dosis de 0,035 mg/kg de peso/día (1mg/m2 de superficie corporal/ día) hasta alcanzar la altura final. El tratamiento deberá interrumpirse después del primer año de tratamiento si la DE de velocidad de crecimiento está por debajo de +1. El tratamiento deberá interrumpirse si la velocidad de crecimiento es de < 2 cm/año y, en caso de necesitar confirmación, la edad ósea es de > 14 años (niñas) o > 16 años (niños), correspondiente al cierre de las placas de crecimiento epifisiario. Talla baja idiopática: Como regla general, se recomienda una dosis de hasta 0,47 mg/kg de peso corporal/semana. La dosis semanal deberá dividirse en 6 ó 7 inyecciones subcutáneas. Posología en pacientes adultos: Deficiencia de hormona de crecimiento (DHC) en adultos La terapia deberá comenzar con una dosis baja, 0,15-0,3 mg por día. La dosis debe ser aumentada gradualmente según las necesidades individuales del paciente, determinada de acuerdo con la concentración del factor de crecimiento I tipo insulina (IGF-I). El fin del tratamiento debería alcanzar concentraciones de IGF-I dentro de 2 SDS de la edad media corregida. Los pacientes con concentración normal de IGF-I al comienzo del tratamiento deberán recibir hormona de crecimiento hasta alcanzar un nivel de IGF-I dentro del rango superior normal, sin exceder de 1,0 mg por día. Las mujeres pueden necesitar dosis más elevadas que los hombres, en aquellos que muestren un aumento de sensibilidad IGF-I en el tiempo. Esto significa que existe el riesgo de que las mujeres, especialmente aquellas que estén recibiendo terapia de sustitución estrogénica oral, estén infra-dosificadas, mientras que los hombres estén sobredosificados. Por tanto, la precisión de dosificación de hormona de crecimiento deberá ser controlada cada 6 mese. Dado que la producción de hormona de crecimiento fisiológica disminuye con la edad, las dosis requeridas de Genotropin pueden reducirse. Se deberá utilizar la dosis mínima eficaz. Preparación y administración: Los viales de GENOTROPIN de 5,3 mg y 12 mg están codificados por color para ayudar a garantizar el uso adecuado con el dispositivo de administración de GENOTROPIN Pen. El vial de 5,3 mg tiene una punta celeste, mientras que el cartucho de 12 mg tiene una punta lila. Los medicamentos parenterales deberían someterse siempre a una inspección visual para descartar la presencia de partículas y de cambios de color antes de su administración, siempre que la solución y el envase lo permitan. GENOTROPIN No debe inyectarse si la solución está turbia o si contiene partículas. Utilizar sólo si es transparente e incoloro. GENOTROPIN puede administrarse en: muslos, glúteos y abdomen, el sitio de las inyecciones SC debe alternarse para evitar lipoatrofia.
Contraindicaciones.
La somatropina está contraindicada en pacientes que tienen evidencia de actividad neoplásica y en pacientes con crecimiento no controlado de tumores intracraneales benignos. La terapia antitumoral se debe llevar a término antes de comenzar la somatropina. La somatropina está contraindicada en pacientes que tienen enfermedades críticas agudas debidas a complicaciones de cirugía de corazón abierto o abdominal, politraumatismo o insuficiencia respiratoria aguda. Dos estudios clínicos controlados con placebo (N=522), realizados en pacientes adultos para evaluar los efectos de la somatropina 5,3 o 8 mg (16 o 24 UI) sobre la duración de la estadía en unidades de cuidados intensivos, demostraron una mortalidad significativamente más alta (41,9% frente a 19,3%) en pacientes tratados con somatropina comparados con los que recibieron placebo. Hipersensibilidad a la sustancia activa o a cualquiera de los excipientes.
Reacciones adversas.
Clases de sistema órgano: Los pacientes que tienen deficiencia de hormona de crecimiento se caracterizan por déficit del volumen extracelular. Cuando se inicia el tratamiento con somatropina, este déficit se corrige rápidamente. En pacientes adultos son frecuentes los efectos adversos relacionados con la retención de fluidos como edema periférico, rigidez musculoesquelética, artralgias, mialgias y parestesias ( > 1/100 y < 1/10). En general, estos efectos adversos son leves a moderados, surgen dentro de los primeros meses de tratamiento y ceden espontáneamente o con la reducción de la dosis. La incidencia de estos efectos adversos está relacionada con la dosis administrada, la edad de los pacientes y quizá inversamente relacionada con la edad de los pacientes al comienzo de la deficiencia de hormona de crecimiento. En los niños estos efectos adversos son poco frecuentes (≥1/1000 y < 1/100): Lista tabulada de reacciones adversas; Las tablas 2-8 muestran las reacciones adversas clasificadas según sistema de órganos y frecuencia usando la siguiente convención: muy frecuente (≥1/10); frecuente (≥1/100 a < 1/10); Infrecuente (≥1/1000 a < 1/100); Raro (≥1/10000 a < 1/1000); muy raro ( < 1/10000); desconocido (no puede ser estimado a partir de los datos disponibles) para cada condición indicada. Estudios clínicos en niños con Deficiencia en la Hormona de Crecimiento: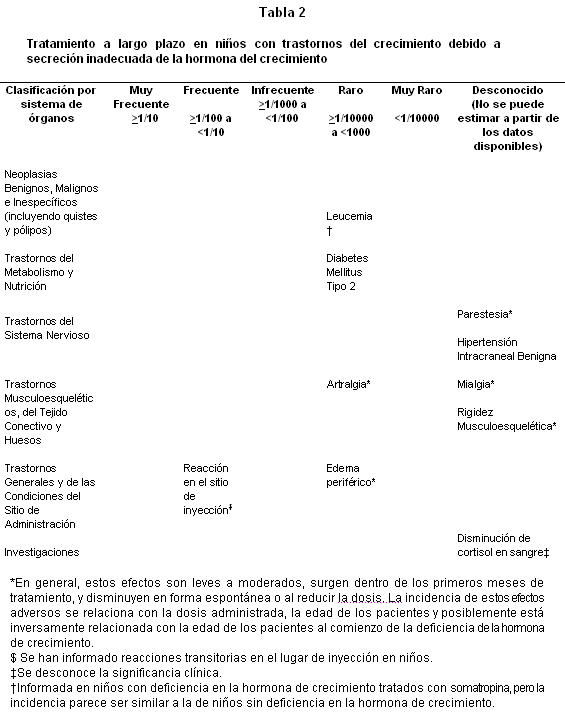
Estudios clínicos en niños con Síndrome Turner: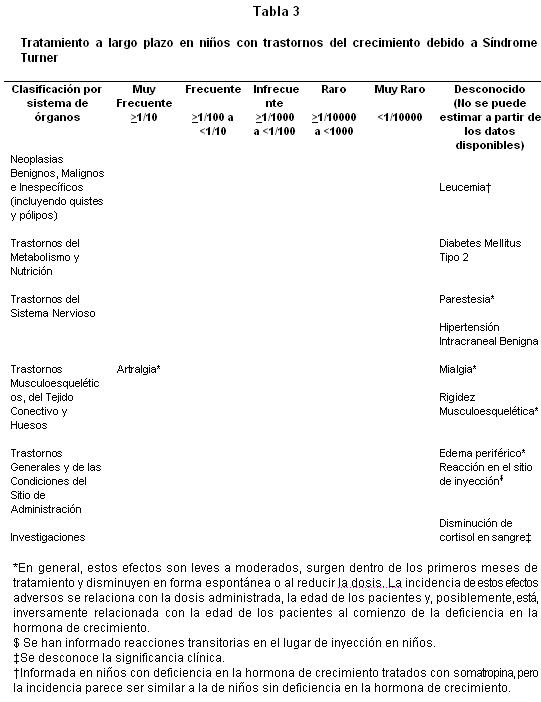
Estudios clínicos en niños con Insuficiencia Renal Crónica: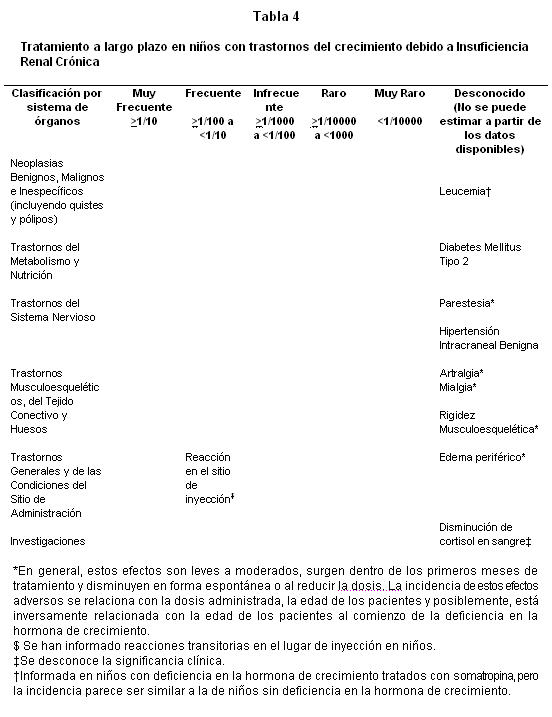
Estudios clínicos en niños con PEG: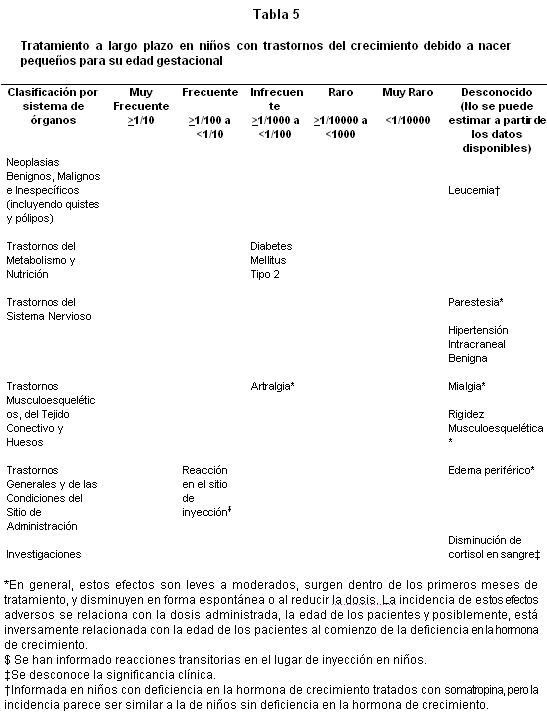
Estudios clínicos en niños con SPW: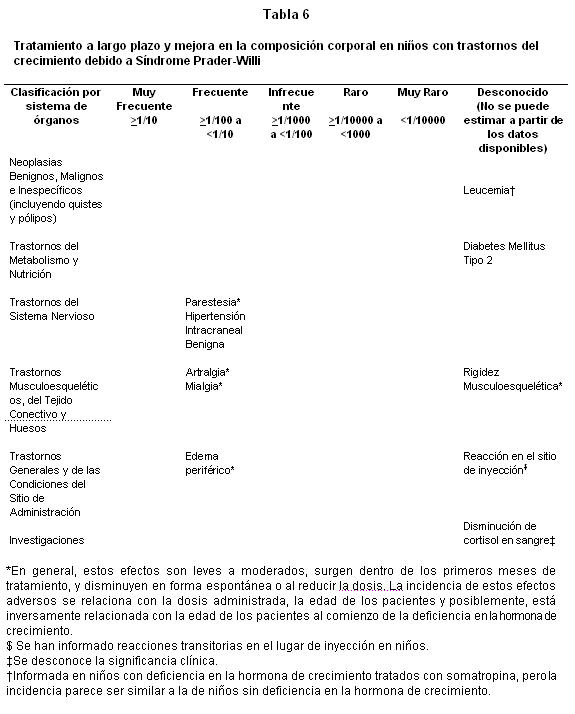
Estudios clínicos en niños con TBI: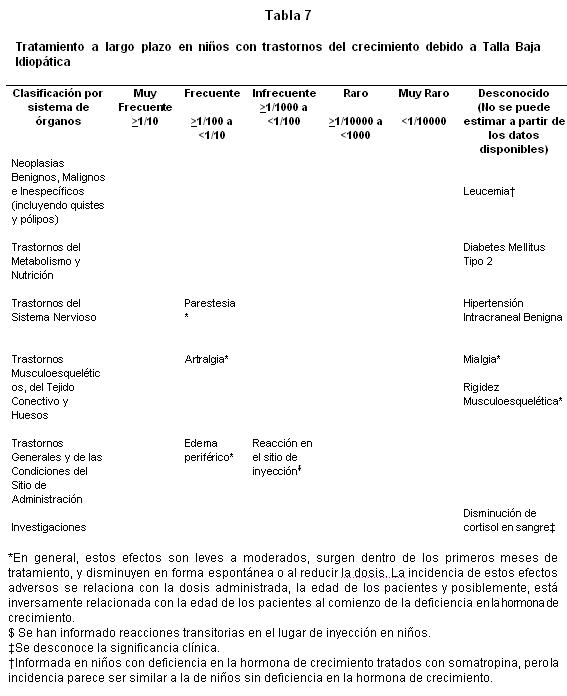
Estudios clínicos en adultos con Deficiencia en la Hormona del Crecimiento: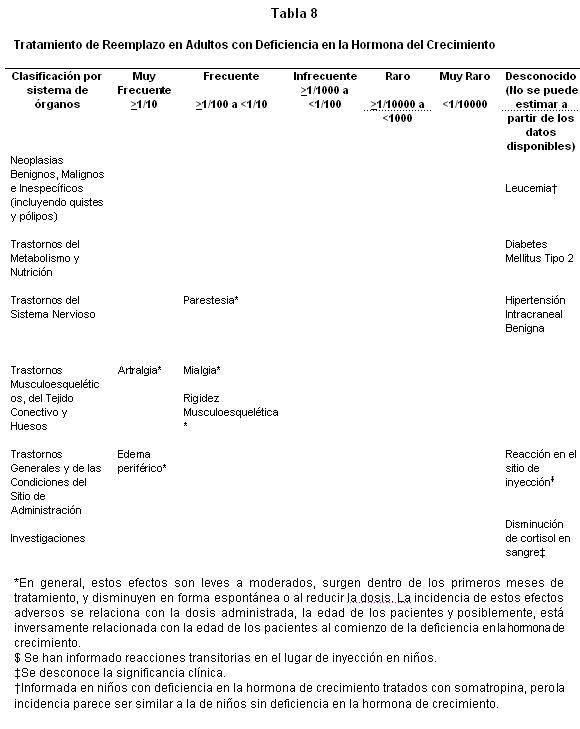
Las reacciones cutáneas transitorias locales al variar el sitio de la inyección en niños son frecuentes (≥1/100 y < 1/10). Se ha informado de unos pocos casos ( < 1/1000 y ≥1/10.000) de hipertensión intracraneal benigna y diabetes mellitus tipo 2. Se ha informado que la somatropina reduce niveles séricos de cortisol. Se desconoce el significado clínico. Se ha informado de unos muy pocos casos ( < 1/10.000) de leucemia en niños con deficiencia de hormona de crecimiento tratados con somatropina, pero la incidencia parece ser similar a la de los niños que no tienen deficiencia de hormona de crecimiento. En la experiencia posterior a la comercialización se ha informado de unos pocos casos de muerte súbita en pacientes afectados por el síndrome de Prader-Willi tratados con somatropina, aunque no se ha demostrado una relación de causalidad.
Advertencias.
Se han producido informes de desenlaces mortales asociados con el uso de hormona de crecimiento en niños aquejados por síndrome de Prader-Willi con uno o más de los siguientes factores de riesgo: obesidad severa, antecedentes de compromiso respiratorio o apnea del sueño o infección respiratoria no identificada. Otro posible factor de riesgo puede ser el género masculino. En los pacientes que tienen síndrome de Prader-Willi se debería evaluar si hay obstrucción de las vías aéreas superiores antes de la iniciación del tratamiento con somatropina. Si durante el tratamiento con somatropina los pacientes muestran signos de obstrucción de las vías aéreas superiores (incluida la aparición o empeoramiento de los ronquidos), se debería suspender el tratamiento. Todos los pacientes que tienen síndrome de Prader-Willi deberían ser evaluados en busca de apnea del sueño y vigilarlos si se sospecha la presencia de esta patología. Estos pacientes también deberían tener un control efectivo del peso y vigilancia de signos de infecciones respiratorias, las cuales se deberían diagnosticar lo más pronto posible y tratarlas de forma radical. La miositis es un evento adverso muy infrecuente que puede estar relacionado con el preservante m-cresol. Si aparecen mialgias o dolor desproporcionado en el lugar de la inyección, se debería considerar el diagnóstico de miositis y, si se confirma, se debería usar una presentación de somatropina que no tenga m-cresol. La somatropina reduce la sensibilidad a la insulina, razón por la cual los pacientes se deberían observar en busca de evidencias de intolerancia a la glucosa. En casos raros, la terapia con somatropina puede producir suficiente intolerancia a la glucosa para cumplir los criterios diagnósticos de una diabetes mellitus tipo 2. El riesgo de que sobrevenga una diabetes durante el tratamiento con somatropina es mayor en aquellos pacientes que tienen otros factores de riesgo para diabetes mellitus tipo 2, como obesidad, antecedentes familiares de diabetes, tratamiento con esteroides o deterioro previo de la tolerancia a la glucosa. En pacientes que tienen diabetes mellitus preexistente es posible que se requiera ajustar la dosis de la terapia antidiabética cuando se instaura el uso de la somatropina. En general, los niveles de la hormona tiroidea periférica permanecen dentro del intervalo de referencia normal durante el tratamiento con somatropina. Sin embargo, se presenta un aumento de la conversión de T4 a T3 que puede dar lugar a una reducción de la T4 sérica y un aumento de las concentraciones séricas de T3. Este efecto puede revestir importancia clínica en pacientes que tienen hipotiroidismo central subclínico en quienes teóricamente puede sobrevenir un hipotiroidismo. A la inversa, puede haber un hipertiroidismo leve en pacientes que reciben terapia de suplencia con tiroxina. Por tal razón, es aconsejable examinar la función tiroidea poco después del comienzo del tratamiento con somatropina y después de hacer ajustes de la dosificación. En pacientes que tienen deficiencia de hormona de crecimiento secundaria por tratamiento de una enfermedad maligna, se recomienda vigilar la aparición de signos de recaída de la enfermedad maligna. En pacientes que tienen trastornos endocrinos, incluida la deficiencia de la hormona de crecimiento, el deslizamiento de las epífisis de la cadera se puede presentar con mayor frecuencia que en la población general. Los niños que comienzan a cojear durante el tratamiento con somatropina deberían ser evaluados. En caso de cefalea grave o recurrente, problemas visuales, náuseas o vómito, se recomienda practicar un examen de fondo de ojo para detectar el papiledema. Si se confirma el papiledema, se debería considerar el diagnóstico de hipertensión intracraneal benigna y, si es del caso, se deberá suspender el tratamiento con hormona de crecimiento. En el presente resulta insuficiente la evidencia para guiar la decisión de si volver o no a introducir la terapia con hormona de crecimiento en pacientes en quienes la hipertensión intracraneal se ha resuelto. Si el tratamiento con hormona de crecimiento se reinicia, se debe realizar una estrecha vigilancia de los síntomas de hipertensión intracraneal. Se puede presentar un avance de la escoliosis en pacientes que experimentan trastornos de crecimiento rápido. Dado que la hormona de crecimiento aumenta la tasa de trastornos del crecimiento, los médicos deben permanecer atentos a esta anormalidad, la cual se puede manifestar durante la terapia con hormona de crecimiento. La escoliosis es frecuente en pacientes que tienen síndrome de Prader-Willi. En pacientes que tienen insuficiencia renal crónica, la función renal debería estar por debajo de 50% de lo normal antes de la institución de la terapia con somatropina. A fin de verificar la presencia de trastornos del crecimiento, estos pacientes deben ser seguidos durante un año antes de la institución de la terapia. Se debe haber establecido el tratamiento conservador de la insuficiencia renal y mantener durante la terapia con hormona de crecimiento. La somatropina se debe suspender cuando se realiza un trasplante renal. Si los pacientes que están recibiendo terapia de suplencia con hormona de crecimiento sufren una enfermedad aguda crítica, el posible beneficio de continuar con el tratamiento con somatropina se debería sopesar contra el riesgo potencial. La somatropina es ineficaz para tratar trastornos del crecimiento en niños que tienen cerradas las epífisis. Fertilidad, Embarazo y lactancia: Los estudios de reproducción en animales no han arrojado evidencias de efectos deletéreos sobre el feto. Sin embargo, no se han hecho estudios en mujeres embarazadas. Dado que los estudios de reproducción en animales no siempre son predictivos de la respuesta humana, la somatropina sólo se debería usar durante el embarazo si la necesidad es clara. En el embarazo normal los niveles de hormona de crecimiento hipofisiaria caen de forma marcada después de la semana 20 de la gestación, siendo reemplazada casi completamente por la hormona de crecimiento placentaria hacia la semana 30. En consecuencia, es poco probable que se necesite continuar la terapia de suplencia con somatropina en mujeres que tienen deficiencia de la hormona de crecimiento durante el tercer trimestre del embarazo. No se sabe si la somatropina se excreta por la leche materna, pero la absorción de proteína intacta en el tracto gastrointestinal del bebé es sumamente improbable.
Interacciones.
La administración de somatropina puede aumentar la depuración de compuestos metabolizados por el citocromo P4503A4 (p.ej., los esteroides sexuales, los corticosteroides, los anticonvulsionantes y la ciclosporina). Se desconoce el significado clínico de esta interacción potencial.
Sobredosificación.
La sobredosis aguda podría llevar inicialmente a hipoglucemia y luego a hiperglucemia. La sobredosis a largo plazo podría redundar en signos y síntomas concordantes con los efectos del exceso de la hormona de crecimiento humana.
Presentación.
GENOTROPIN Solución inyectable de 16 U.I./mL Cartridge x 1 Ud. GENOTROPIN Solución inyectable de 36 U.I./mL Cartridge x 1 Ud.