HEPTA
MEGALABS
Factor hormonal estimulante de precursores de la eritropoyesis.
Descripción.
La Eritropoyetina es una glicoproteína que estimula la producción de glóbulos rojos. Se produce en el riñón y estimula la división y diferenciación de las células eritroides que se encuentran en la médula ósea. HEPTA es una Eritropoyetina alfa humana recombinante (r-hu-Eritropoyetina), una glicoproteína de 165 aminoácidos producida por tecnología de ADN recombinante, y presenta los mismos efectos endócrinos que la eritropoyetina endógena. Tiene un peso molecular de 30.400 Daltons y la misma secuencia de aminoácidos que la eritropoyetina natural.
Composición.
Cada frasco ampolla o jeringa prellenada contiene: HEPTA (r-Hu Eritropoyetina alfa) 2000 UI: r-Hu Eritropoyetina alfa 2000 Ul. Cloruro de Sodio 5,84 mg. Fosfato monosódico dihidrato 1,164 mg. Fosfato disódico dihidrato 2,225 mg. Albúmina humana 20% 2,50 mg. Agua para inyección c.s.p. 1,00 ml. HEPTA (r-Hu Eritropoyetina alfa) 4000 UI: r-Hu Eritropoyetina alfa 4000 Ul. Cloruro de Sodio 5,84 mg. Fosfato monosódico dihidrato 1,164 mg. Fosfato disódico dihidrato 2,225 mg. Albúmina humana 20% 2,50 mg. Agua para inyección c.s.p. 1,00 ml.
Farmacología.
Mecanismo de acción: El gen de la eritropoyetina se ha clonado y ahora se encuentra disponible para el tratamiento de la anemia debida a su deficiencia. La eritropoyetina es una hormona producida por el riñón que promueve la proliferación y maduración de progenitores eritroides. Esto se traduce en un aumento del recuento de reticulocitos, seguido por una elevación de la hemoglobina y del hematocrito. Farmacocinética: Luego de una administración IV, la eritropoyetina tiene una eliminación que sigue una cinética de primer orden, siendo la vida media entre 4 y 13 horas en el adulto y niño con insuficiencia renal crónica (IRC). Dentro del rango de dosis terapéutico, se mantienen niveles de eritropoyetina detectables en plasma durante al menos 24 horas. Luego de la administración subcutánea de eritropoyetina a pacientes con IRC, el peak sérico se alcanza a las 5-24 horas luego de la administración y declina lentamente de allí en adelante. No hubo diferencias aparentes en la vida media entre adultos fuera de diálisis con niveles de creatinina mayores de 3 y pacientes adultos en diálisis. En voluntarios normales, la vida media intravenosa de eritropoyetina es aproximadamente un 20% más corta que la vida media en pacientes con IRC. La farmacocinética de eritropoyetina no se ha estudiado en pacientes infectados por HIV. El perfil farmacocinético de eritropoyetina en niños y adolescentes parece ser similar al de los adultos. Hay datos limitados en neonatos. Al comienzo de la acción hay un incremento del recuento de reticulocitos (efecto inicial) dentro de los 7 a 10 días posteriores a la administración. Generalmente en 2 a 6 semanas ocurren aumentos en el recuento de glóbulos rojos, hematocrito y hemoglobina, clínicamente significativos. La velocidad y el grado de respuesta son dependientes de la dosis y de la disponibilidad de hierro de los depósitos. En un periodo de 2 semanas, la administración de 50 unidades por kg de peso tres veces por semana, aumenta el hematocrito en 1,5 puntos. La administración de 100 unidades por kg de peso tres veces por semana aumenta el hematocrito en 2,5 puntos y la administración de 150 unidades por kg de peso tres veces por semana lo aumenta 3,5 puntos.
Toxicología.
Toxicología Animal: Determinación de la DL 50 l/v en perros: 1000 UI/Kg/día. Determinación de la DL 50 l/v en ratas: 1000 UI/Kg/día. Se administró R-HU-Eritropoyetina alfa IV durante 4 días consecutivos en perros. Las dosis fueron: 0.20, 200 y 500 UI/Kg/día. Se estudió la presión sanguínea, la frecuencia respiratoria y cardiaca antes de la administración, en forma horaria y cada 5 horas después de la infusión de la droga durante 4 días. También se controló el peso corporal y la ingesta de comida. No se reportaron efectos adversos con ninguna dosis en los perros incluso con las dosis más altas administradas, que representan el 25% de la dosis administrada en humanos. Estudio de toxicidad sub aguda: Se administraron dosis repetidas de R-HU-eritropoyetina ALFA a ratas Sprague-Dawley ( > 400 gr de peso) con nefrectomía y a un grupo control en ratas sanas. Las ratas recibieron 6 UI en 1 ml SC durante 33 días. El examen histopatológico de los nódulos linfáticos, bazo e hígado no reveló producción extra medular de glóbulos rojos. El examen inmuno histológico de los riñones no mostró acumulación de inmunoglobulina. El examen de los pulmones mostró embolias en las ratas que desarrollaron IRC. Toxicología reproductiva: Carcinogenesis, Mutagenesis, y deterioro de la fertilidad: No se evaluó la carcinogénesis potencial de la eritropoyetina, pero no induce mutación génica bacteriana (Test de Ames), aberraciones cromosómicas en células de mamíferos, micronúcleo de ratón o mutación genética en el locus HGPRT. En ratas hembras tratadas con eritropoyetina IV, hubo una tendencia leve a la pérdida fetal a la dosis de 100 y 500 Unidades/kg.
Indicaciones.
HEPTA está indicado para: Tratamiento de la anemia en pacientes con falla renal crónica: La eritropoyetina está indicada en el tratamiento de la anemia asociada a falla renal crónica, incluyendo pacientes en diálisis (etapa terminal de la insuficiencia renal crónica) así cómo en pacientes que no están en diálisis. La eritropoyetina se indica para elevar o mantener el nivel de los glóbulos rojos (determinados por el hematocrito y la hemoglobina) y disminuir la necesidad de transfusiones en estos pacientes. Los pacientes que no están en diálisis con anemia sintomática deben tener un hematocrito menor a 30% para considerar la terapia con eritropoyetina. No está indicado el uso de eritropoyetina para pacientes que requieren una corrección inmediata de una anemia severa. El uso de eritropoyetina puede obviar la necesidad de transfusiones sanguíneas de mantenimiento, pero no es un sustituto para la transfusión de emergencia. Previo al inicio de la terapia, deberán valorarse los depósitos de hierro, incluyendo la saturación de transferrina, y ferritina sérica. La saturación de transferrina debe ser al menos del 20% y la ferritina al menos 100 ng/ml. La presión sanguínea debe ser monitorizada de cerca y controlada durante la terapia. Tratamiento de la anemia en los pacientes infectados con el virus de HIV y tratados con Zidovudina: La eritropoyetina está indicada en el tratamiento de la anemia relacionada con la terapia con zidovudina (AZT) en pacientes infectados con el virus del HIV. Se indica para elevar o mantener los niveles de glóbulos rojos (determinados por el hematocrito o las determinaciones de hemoglobina) y reducir las necesidades de transfusión en estos pacientes. La eritropoyetina no está indicada para el tratamiento de la anemia en pacientes HIV positivos debida a otros factores tales como: deficiencia de hierro o folatos, hemólisis o sangrado gastrointestinal. El uso de eritropoyetina, a la dosis de 100 Unidades/kg 3 veces por semana es efectiva para disminuir las necesidades de transfusión y aumentar los niveles de glóbulos rojos en los pacientes anémicos infectados por HIV, tratados con zidovudina, cuando los niveles de eritropoyetina endógena sérica son inferiores a 500 Unidades/ml y la dosis de zidovudina no supera los 4.200 mg/semana. Tratamiento de la anemia en pacientes con cáncer en Quimioterapia: La terapia con eritropoyetina está indicada para el tratamiento de la anemia en pacientes con neoplasias no mieloides, en quiénes la anemia se debe al efecto de la quimioterapia concomitante. La eritropoyetina está indicada para disminuir el número de transfusiones en pacientes que recibirán quimioterapia, durante al menos 2 meses. No está indicada para el tratamiento de la anemia debido a deficiencias de folato o hierro, hemólisis o sangrado gastrointestinal. Reducción de la transfusión sanguínea alogénica en pacientes quirúrgicos: El uso de eritropoyetina está indicado para reducir el riesgo de transfusiones en pacientes anémicos (hemoglobina entre 10 y 13 g/dl) que han sido coordinados para cirugía no cardiaca, no vascular. Su administración está indicada en pacientes con alto riesgo de transfusiones peri-operatorias, en los cuales se anticipa una pérdida de sangre importante. No se indica el uso de eritropoyetina para los pacientes que desean donar sangre autóloga. La seguridad del uso peri-operatorio de eritropoyetina se ha estudiado solamente en pacientes que recibían profilaxis anticoagulante.
Dosificación.
HEPTA puede administrarse por vía intravenosa o subcutánea. En general se administra por vía intravenosa a pacientes con acceso venoso disponible, como en el caso de pacientes que están recibiendo hemodiálisis, y por vía intravenosa o subcutánea a cualquier otro paciente. Pacientes con insuficiencia renal crónica: Las dosis de inicio se encuentran dentro del rango de 50-100 Ul/kg 3 veces por semana. Estas dosis han demostrado ser seguras y efectivas en aumentar el hematocrito y eliminar la dependencia de las transfusiones en pacientes con insuficiencia renal crónica. La dosis de HEPTA debe reducirse cuando el hematocrito se aproxima al 36% o aumenta más de 4 puntos en 2 semanas. HEPTA puede ser administrada por inyección IV o SC. En los pacientes en hemodiálisis la eritropoyetina en general se administra en bolo IV al final del procedimiento dialítico.
Durante el tratamiento se recomienda controlar los parámetros hematológicos en forma regular. Evaluación pre-tratamiento del hierro: Previo y durante el tratamiento con eritropoyetina deben evaluarse los depósitos de hierro, incluyendo la saturación de transferrina y la ferritina sérica. La saturación de transferrina debe ser al menos del 20%, y la ferritina de al menos 100 ng/ml. En general todos los pacientes requerirán suplementación con hierro para aumentar o mantener la saturación de transferrina, lo que sustentará adecuadamente la eritropoyesis inducida por la eritropoyetina. Ajuste de dosis: Se requiere un período de tiempo para que los progenitores eritroides maduren y sean liberados a la circulación. Además, la vida media de los glóbulos rojos puede variar debido a la uremia. Esto determina que el período de tiempo requerido para alcanzar un cambio clínico en el hematocrito (aumento o descenso) para cualquier ajuste de dosis sea de 2 a 6 semanas. Los ajustes no deben ser más frecuentes que una vez por mes, a menos que exista una indicación clínica. Después de un ajuste de dosis, el hematocrito deberá ser determinado en forma semanal durante al menos 2 a 6 semanas. Si el nivel de hematocrito aumenta y se acerca al 36%, la dosis deberá reducirse para mantener el hematocrito dentro del rango sugerido. Si este descenso de dosis no detiene la elevación del hematocrito y éste excede el 36% la dosis deberá ser suspendida en forma temporal hasta que el hematocrito descienda, comenzando nuevamente la terapia a dosis menores. En cualquier momento, si el hematocrito aumenta en más de 4 puntos en 2 semanas, la dosis deberá descenderse inmediatamente. Luego de la reducción de la dosis, se monitorizará el hematocrito 2 veces por semana y se evaluará la necesidad de nuevos ajustes de dosis, tal cómo se menciona en el apartado DOSIS DE MANTENIMIENTO. Si el incremento del hematocrito no es de 5-6 puntos en un período de 8 semanas y los depósitos de hierro son adecuados, la dosis de eritropoyetina puede incrementarse progresivamente. Podrán realizarse más incrementos en intervalos de 4-6 semanas hasta alcanzar la respuesta deseada. Dosis de mantenimiento: La dosis de mantenimiento deberá individualizarse para cada paciente en diálisis. Si el hematocrito permanece bajo o por debajo del rango sugerido, deberán re-evaluarse los depósitos de hierro. Si la saturación de transferrina es menor al 20% se administrarán suplementos de hierro. Si la saturación de transferrina es mayor al 20%, la dosis de eritropoyetina puede ser incrementada. Estos aumentos de dosis no deben realizarse con una frecuencia mayor a una vez por mes, a menos que esté clínicamente indicado. El hematocrito deberá determinarse en forma semanal por las siguientes 2-6 semanas después de un aumento de dosis. En pacientes con insuficiencia renal crónica en diálisis, la dosis de mantenimiento deberá individualizarse. Dosis de eritropoyetina de 75-150 Ul/kg por semana han mostrado mantener el hematocrito entre 36-38% hasta por 6 meses. Falta de respuesta o ausencia de respuesta: Cerca del 95% de los pacientes con insuficiencia renal crónica respondieron con incrementos clínicamente significativos del hematocrito y casi todos fueron independientes de las transfusiones en un plazo de aproximadamente 2 meses después de iniciada la terapia con eritropoyetina. Si un paciente no responde o no se logra mantener la respuesta, deberán considerarse otras causas de falta de respuesta. Pacientes infectados por el virus del HIV tratados con zidovudina: Antes de comenzar la terapia con HEPTA, se recomienda determinar los niveles endógenos de eritropoyetina sérica (antes de realizar transfusiones). La evidencia disponible sugiere que los pacientes que reciben zidovudina con niveles de eritropoyetina sérica mayores a 500 Ul/ml pueden no responder a la terapia con eritropoyetina. Dosis de Inicio: Para los pacientes con eritropoyetina sérica ≤ 500 Ul/ml que reciben dosis de zidovudina no mayores a 4200 mg/semana, la dosis de inicio recomendada es de 100 Ul/kg IV o SC 3 veces por semana, por 8 semanas. Aumento de dosis: Durante el ajuste de la dosis, el hematocrito debe ser monitorizado en forma semanal. Si la respuesta no es satisfactoria en cuanto a la reducción de la necesidad de transfusiones o al aumento deseado del nivel de hematocrito después de 8 semanas de tratamiento, la dosis de HEPTA puede ser aumentada en 50-100 Ul/kg 3 veces por semana. La respuesta debe ser evaluada cada 4 a 8 semanas. Si el paciente no responde satisfactoriamente a dosis de HEPTA de 300 Ul/kg, 3 veces por semana es poco probable que responda a dosis mayores de la droga. Dosis de mantenimiento: Luego de alcanzar la respuesta deseada (reducción de los requerimientos de transfusión o aumento del hematocrito) la dosis de HEPTA debe ser individualizada para mantener la respuesta basándose en factores tales como la dosis de zidovudina y la presencia de infecciones intercurrentes o episodios de inflamación. Si el hematocrito excede el 40% deberá discontinuarse la droga hasta que caiga al 36%. La dosis deberá reducirse en un 25% cuando el tratamiento se reinicie y luego titulada para mantener el nivel de hematocrito deseado. Pacientes con cáncer en quimioterapia: En general, los pacientes con niveles basales séricos de eritropoyetina más bajos responden más vigorosamente a la administración de HEPTA que los pacientes con niveles más altos. Aunque no se puede estipular el nivel sérico de eritropoyetina por encima del cual es poco probable que los pacientes responden favorablemente a la terapia con HEPTA, no se recomienda la administración de la droga a pacientes con niveles de eritropoyetina sérica elevados (ejemplo > 200 UI/ml). Se controlará el hematocrito en forma semanal en estos pacientes hasta que el mismo se estabilice. La dosis de inicio recomendada es de 150 Ul/kg SC, 3 veces por semana. Si la respuesta no es satisfactoria en términos de reducción de requerimiento de transfusión o aumento de hematocrito en 8 semanas de terapia la dosis de HEPTA puede ser aumentada hasta 300 Ul/kg 3 veces por semana. Si el hematocrito excede el 40% la dosis de eritropoyetina deberá ser suspendida hasta que el hematocrito caiga por debajo del 36%. Si la dosis inicial de HEPTA provoca una respuesta muy rápida del hematocrito (aumento de más de 4 puntos porcentuales en 2 semanas de tratamiento) la dosis deberá ser reducida. Pacientes quirúrgicos: Antes de comenzar la terapia con eritropoyetina, la hemoglobina debe estar entre 10 y 13 g/dI. La dosis de HEPTA recomendada es de 300 Ul/kg/día SC 10 días antes de la cirugía, el día de la cirugía y hasta 4 días después de la misma. Un esquema alternativo es 600 Ul/kg de HEPTA SC en una dosis semanal (21, 14, y 7 días antes de la cirugía) más una cuarta dosis el día de la operación. Todos los pacientes deben recibir una suplementación adecuada de hierro. El aporte de hierro deberá iniciarse al comienzo de la terapia con HEPTA y continuar durante todo el tratamiento.
Contraindicaciones.
Hipertensión arterial no controlada. Hipersensibilidad reconocida a algunos de los componentes del producto.
Reacciones adversas.
Inmunogenicidad: Al igual que lo que sucede con todas las proteínas que se utilizan en forma terapéutica existe la posibilidad de desarrollar inmunogenicidad. Se han reportado algunos casos de APCR asociados a anticuerpos neutralizantes con el uso de eritropoyetina recombinante. Estos casos se observaron en pacientes tratados tanto por vía SC cómo IV y predominantemente en pacientes con IRC. Pacientes con falla renal crónica: El análisis de los estudios indica, en general, que la eritropoyetina es bien tolerada. Los efectos adversos reportados son frecuentemente secuela de la falla renal crónica y no se pueden atribuir necesariamente a la terapia con eritropoyetina.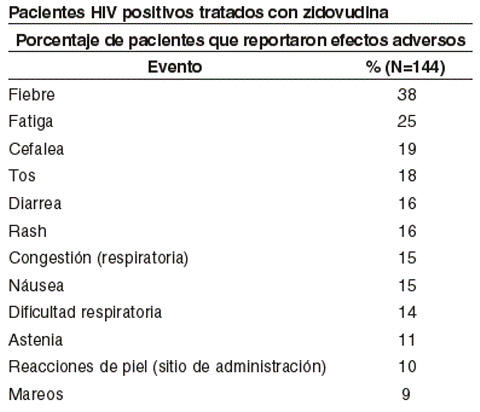
Los eventos adversos que ocurrieron dentro de las primeras horas después de la administración de eritropoyetina fueron raros, moderados y transitorios incluyendo reacción local en el sitio de inyección en pacientes en diálisis y síntomas seudo-gripales cómo artralgias y mialgias. Hipertensión: Se han reportado aumentos de la presión arterial en algunos estudios clínicos en general durante los primeros 90 días de tratamiento. En forma ocasional se observó encefalopatía hipertensiva y convulsiones en pacientes con IRC en tratamiento con eritropoyetina. Hubo una tendencia a presentar mayor probabilidad de eventos adversos hipertensivos en pacientes que presentaban incrementos más rápidos del hematocrito (mayor a 4 puntos porcentuales en 2 semanas). Convulsiones: Parece haber una tasa mayor de convulsiones durante los primeros 90 días de terapia (en aproximadamente el 2,5% de los pacientes) cuando se compara con períodos subsiguientes de 90 días. La incidencia basal de convulsiones en la población en diálisis no tratada es difícil de determinar, pero parece estar en el rango del 5% al 10% por pacientes por año. Reacciones alérgicas: No se observaron reacciones alérgicas o anafilácticas serias. Se observa en general rash o urticaria, aunque poco frecuente, y moderados y transitorios. Pacientes HIV positivos tratados con Zidovudina: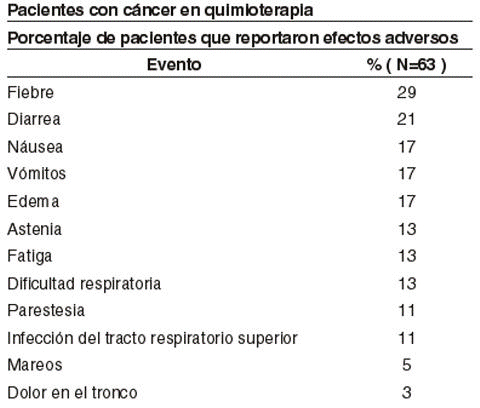
Reacciones alérgicas: Se han reportado reacciones tipo urticaria dentro de las 48 horas de la exposición a la medicación. Convulsiones: Han existido reportes de convulsiones en el tratamiento de pacientes con HIV que recibían eritropoyetina y zidovudina. Estas reacciones parecen relacionarse con patología subyacente como meningitis o neoplasmas cerebrales, no a la terapia con eritropoyetina. Pacientes con cáncer en quimioterapia:
Pacientes quirúrgicos:
Eventos trombótlcos: En estudios clínicos, la frecuencia de trombosis venosa profunda fue similar en los pacientes tratados con eritropoyetina y en el grupo placebo. Carcinogenesis, mutagenesis y trastorno de la fertilidad: El potencial carcinogénico de eritropoyetina no ha sido evaluado. El uso de eritropoyetina no induce mutación génica en bacterias (Test de Ames), aberraciones cromosómicas en células de mamíferos, micronúcleo de ratón o mutación genética en el locus HGPRT. En ratas hembras tratadas IV con eritropoyetina hubo una leve tendencia a la pérdida fetal a la dosis de 100 y 500 Ul/kg. Embarazo v lactancia: No hay estudios bien controlados en mujeres embarazadas. Debería reservarse el uso de eritropoyetina durante el embarazo para los casos en los cuales el beneficio potencial justifica el riesgo para el feto. Se desconoce si la eritropoyetina se excreta en la leche humana. Dado que muchas drogas se eliminan por esta vía deberá tenerse precaución cuando la droga se administra a una mujer en lactancia. Uso en pediatría: La seguridad y eficacia de eritropoyetina en niños no ha sido establecida.
Precauciones.
La administración de cualquier producto biológico por vía parenteral debe ser controlada de cerca para evaluar las posibles reacciones alérgicas u otro efecto secundario. En los estudios clínicos se observó en forma ocasional la aparición de rash, no se reportó ninguna reacción alérgica o anafiláctica. La seguridad y eficacia de eritropoyetina no se ha establecido en pacientes con historia conocida de enfermedades hematológicas subyacentes con anemia, síndromes mielodisplásicos o hipercoagulación. Hematología: Se ha observado la exacerbación de la porfiria en forma ocasional en pacientes tratados con eritropoyetina portadores de enfermedad renal crónica. Sin embargo, el uso de eritropoyetina no causó incremento en la eliminación urinaria de metabolitos porfirínicos en voluntarios sanos. En estudios preclínicos en ratas y perros la terapia con eritropoyetina se asoció con fibrosis subclínica de la médula ósea. La fibrosis de la médula ósea es una complicación conocida de la falla renal crónica en humanos y podría relacionarse con el hiperparatiroidismo secundario u otros factores no conocidos. La incidencia de fibrosis de médula ósea no aumentó en un estudio en pacientes en diálisis tratados con eritropoyetina por 12 a 19 semanas. En pacientes con falla renal crónica debe controlarse el hematocrito 2 veces por semana; en los pacientes infectados con el virus del HIV tratados con zidovudina y en los pacientes con cáncer, los niveles de hematocrito deben controlarse 1 vez por semana hasta que se estabilice el hematocrito y luego en forma periódica. Respuesta demorada o disminuida: Si el paciente no responde o no logra mantener la respuesta dentro del rango recomendado con la dosis administrada deberán considerarse y evaluarse las siguientes etiologías: 1. Deficiencia de hierro: generalmente todos los pacientes requieren aporte de hierro suplementario. 2. Infecciones subyacentes, procesos inflamatorios o malignos. 3. Pérdida oculta de sangre. 4. Enfermedades hematológicas subyacentes (talasemia, anemia refractaria). 5. Déficit de vitaminas: ácido fólico o vitamina B12. 6. Hemólisis. 7. Intoxicación por aluminio. 8. Osteítis fibrosa cística. 9. Aplasia pura de células rojas (APCR): en ausencia de otra etiología, el paciente debe ser evaluado en busca de APCR y se debe realizar la investigación en suero de anticuerpos contra eritropoyetina recombinante. Evaluación férrica: Durante el tratamiento con HEPTA puede desarrollarse un déficit absoluto o funcional de hierro. El déficit funcional de hierro, con niveles normales de ferritina, pero baja saturación de transferrina, se debe presumiblemente a la imposibilidad de movilizar los depósitos de hierro lo suficientemente rápido como para mantener la eritropoyesis aumentada. La saturación de transferrina debe ser al menos de 20% y la ferritina de al menos 100 ng/ml. Antes y durante la terapia con HEPTA deberá evaluarse el estatus férrico del paciente con determinación de la saturación de transferrina y ferritina sérica. En general todos los pacientes requieren de la suplementación de hierro para incrementar o mantener los niveles de saturación de transferrina que mantendrán la eritropoyesis estimulada por eritropoyetina.
Advertencias.
Falla renal crónica e hipertenslón: Los pacientes portadores de hipertensión arterial no controlada no deberían ser tratados con eritropoyetina; la presión arterial debe controlarse adecuadamente antes de iniciar la terapia. Hasta un 80% de los pacientes con falla renal crónica tiene historia de hipertensión. Aunque no parece haber efectos presores directos de la eritropoyetina, la presión arterial puede aumentar durante la terapia con HEPTA. Durante la fase precoz del tratamiento hasta un 25% de los pacientes en diálisis pueden requerir terapia antihipertensiva. En pacientes con insuficiencia renal crónica tratados con eritropoyetina se ha observado la presencia de encefalopatía hipertensiva. Deberá prestarse especial atención y monitorizar de cerca la presión arteriaI y realizar un tratamiento agresivo de la misma. Se les advertirá a los pacientes la importancia del cumplimiento de la terapia antihipertensiva y la restricción dietética. Se recomienda disminuir la dosis de eritropoyetina si el incremento del hematocrito excede los 4 puntos porcentuales en 2 semanas, debido a la posible asociación entre el aumento excesivo del hematocrito con la exacerbación de la hipertensión. En pacientes con insuficiencia renal crónica en hemodiálisis con evidencia clínica de isquemia cardiaca o falla cardiaca congestiva el hematocrito debe controlarse y no exceder el 36%. Convulsiones: En pacientes en diálisis, hubo una mayor incidencia de convulsiones durante los primeros 90 días de la terapia (2.5% de los pacientes). Debido al riesgo potencial de aumento de las convulsiones en los primeros 90 días de terapia, la presión arterial y la presencia de síntomas premonitorios deberán controlarse de cerca. Se le deberá advertir a los pacientes que eviten realizar actividades potencialmente peligrosas, como por ejemplo conducir u operar maquinaria pesada durante este período. La relación entre las convulsiones y el aumento del hematocrito es incierta. Eventos trombótlcos: Durante la hemodiálisis los pacientes tratados con HEPTA pueden requerir un aumento de la anticoagulación para evitar la aparición de eventos trombóticos. Se han reportado otros eventos trombóticos como infarto de miocardio, accidentes vasculares, accidentes isquémicos transitorios. Los pacientes con enfermedad vascular preexistente deberán controlarse de cerca.
Interacciones.
Las siguientes drogas pueden causar interacciones con HEPTA: Drogas antihipertensivas: El uso de HEPTA puede provocar el aumento de la presión arterial, en especial si el hematocrito aumenta muy rápidamente. Puede requerirse la utilización de más terapia antihipertensiva. Heparina: Durante la hemodiálisis los pacientes pueden requerir un aumento de la dosis de heparina para evitar la aparición de fenómenos trombóticos.
Conservación.
Almacenar entre 2° a 8°C (36° a 46°F). No congelar o agitar. Preparación y administración: 1. No agitar. La agitación vigorosa y prolongada puede desnaturalizar cualquier glicoproteína y producir la inactivación biológica. 2. Retire la jeringa o el vial del envase y controle que la solución sea clara, incolora y sin partículas visibles. 5. Proceda a la inyección de la solución IV o SC, siguiendo una técnica de inyección adecuada.
Sobredosificación.
No se ha determinado la cantidad de eritropoyetina que puede ser administrada en forma segura en dosis única o múltiple. Se han administrado dosis de hasta 1,500 Ul/kg 3 veces por semana durante 3 a 4 semanas sin efectos tóxicos directos. El tratamiento con eritropoyetina puede resultar en policitemia si no se controla adecuadamente el hematocrito.
Presentación.
HEPTA 2.000 UI/jeringa prellenada de 1 ml. HEPTA 4.000 UI/jeringa prellenada de 1 ml.