HERCEPTIN®
ROCHE
Agente antineoplásico.
Composición.
Principio activo: trastuzumab. Forma farmacéutica: Viales monodosis con 150 mg y viales multidosis con 440 mg de polvo para concentrado para solución para infusión. El concentrado reconstituido de Herceptin contiene 21 mg/ml de trastuzumab.
Farmacología.
Propiedades farmacodinámicas: Mecanismo de acción: Trastuzumab es un anticuerpo monoclonal humanizado obtenido por técnicas de ADN recombinante, de acción selectiva sobre el dominio extracelular de la proteína del receptor 2 del factor del crecimiento epidérmico humano (HER2). El anticuerpo es una IgG1 que contiene regiones marco humanas con las regiones determinantes de complementariedad de un anticuerpo murino HER2 anti-p185 que se une a HER2. El protooncogén HER2 o c-erbB2 codifica una sola proteína transmembranaria de tipo receptor, de 185kDa y estructuralmente relacionada con el receptor del factor de crecimiento epidérmico. En el 25-30% de los casos de carcinoma de mama primario se observa sobreexpresión de HER2. La amplificación del gen HER2 conlleva una mayor expresión de la proteína HER2 en la superficie de estas células tumorales, lo cual se traduce en receptores HER2 constitutivamente activados. Los estudios muestran que los pacientes con carcinoma de mama cuyos tumores presentan una amplificación o sobreexpresión de HER2 tienen una supervivencia sin enfermedad (SSE) más corta que la de aquellas con tumores sin amplificación o sobreexpresión de HER2. Tanto en los ensayos in vitro como en estudios en animales, se ha demostrado que trastuzumab inhibe la proliferación de las células tumorales humanas que sobreexpresan HER2. También se ha demostrado in vitro que la citotoxicidad dependiente de anticuerpos mediada por células (ADCC) del trastuzumab se ejerce preferentemente sobre las células cancerosas que sobreexpresan HER2 antes que sobre las que no sobreexpresan HER2. Ensayos clínicos / Eficacia: Carcinoma de mama: CMM: En los ensayos clínicos, Herceptin se ha utilizado en monoterapia en pacientes con carcinoma de mama metastásico con sobreexpresión de HER2 que habían recaído tras una o más pautas quimioterápicas contra su enfermedad metastásica. Herceptin también se ha administrado en estudios clínicos en combinación con paclitaxel o una antraciclina (doxorrubicina o epirrubicina) y ciclofosfamida (AC) como tratamiento de primera línea de pacientes con carcinoma de mama metastásico con sobreexpresión de HER2. Pacientes que habían recibido previamente quimioterapia basada en antraciclinas recibieron paclitaxel (175 mg/m2 en infusión de 3 horas) con o sin Herceptin. Se les administró Herceptin hasta la progresión de la enfermedad. Con Herceptin en monoterapia como tratamiento de segunda o tercera línea de mujeres con carcinoma de mama metastásico y sobreexpresión de HER2 se ha obtenido una tasa global de respuesta tumoral del 15% y una mediana de supervivencia de 13 meses. La combinación de Herceptin y paclitaxel como tratamiento de primera línea de mujeres con carcinoma de mama metastásico e sobreexpresión de HER2 prolonga significativamente la mediana del tiempo hasta la progresión en comparación con paclitaxel en monoterapia. La mediana del tiempo hasta la progresión de la enfermedad aumenta 3,9 meses (6,9 frente a 3,0 meses) en los pacientes tratados con paclitaxel. La respuesta tumoral y la tasa de supervivencia al cabo de un año también aumentan con Herceptin en combinación con paclitaxel en comparación con paclitaxel en monoterapia. En un ensayo clínico controlado y aleatorizado se ha estudiado también Herceptin en combinación con docetaxel como tratamiento de primera línea de mujeres con carcinoma de mama metastásico. La biterapia con Herceptin y docetaxel elevó significativamente la tasa de respuesta (61% frente al 34%) y prolongó la mediana de la duración hasta la progresión de la enfermedad (en 5,6 meses) en comparación con los pacientes tratadas con docetaxel solo. En comparación con docetaxel en monoterapia, la mediana de la supervivencia también aumentó significativamente con esta biterapia (31,2 frente a 22,7 meses). Biterapia con Herceptin y anastrozol: Se ha estudiado la biterapia con Herceptin y anastrozol como tratamiento de primera línea del carcinoma de mama metastásico en pacientes con sobreexpresión de HER2 y positividad de los receptores hormonales (es decir, receptores de estrógenos [RE] y de progesterona [RP]). El tiempo de supervivencia sin progresión fue el doble de largo en el grupo con Herceptin + anastrozol que en el de anastrozol solo (4,8 frente a 2,4 meses). Con la biterapia se alcanzaron también mejores resultados en otros parámetros: respuesta global (16,5% frente al 6,7%), beneficio clínico (42,7% frente al 27,9%) y tiempo hasta la progresión (4,8 frente a 2,4 meses). En el tiempo hasta la respuesta y la duración de esta no se observó ninguna diferencia entre ambos grupos. La mediana de la supervivencia global aumentó en 4,6 meses en el grupo con la biterapia. Esta diferencia no era estadísticamente significativa; ahora bien, más de la mitad de los pacientes del grupo con anastrozol en monoterapia cambiaron a la pauta con Herceptin tras la progresión de la enfermedad. El 52% de los pacientes tratadas con Herceptin + anastrozol sobrevivieron durante un mínimo de 2 años frente al 45% de los que recibieron anastrozol en monoterapia. CMP: En el uso adyuvante, Herceptin se ha estudiado en 4 ensayos clínicos aleatorizados y multricéntricos a gran escala: El estudio HERA se diseñó para comparar un año de administración de Herceptin cada tres semanas con solo observación en pacientes que sufrían carcinoma de mama precoz HER2-positivo tras cirugía, quimioterapia convencional y radioterapia (si procedía). A los pacientes del grupo de Herceptin se les administró una dosis de carga de 8 mg/kg, seguida de 6 mg/kg cada tres semanas durante un año. Los estudios NCCTG N9831 y NSAPB B31M, con análisis conjuntos, se diseñaron para evaluar la utilidad clínica de combinar Herceptin con paclitaxel a la quimioterapia con AC. Además, en el estudio NCCTG N9831 se investigó la adición secuencial de Herceptin a la quimioterapia con AC-paclitaxel tras la cirugía en pacientes con carcinoma de mama precoz HER-2 positivo tras la cirugía. El estudio BCTRG 006 se diseñó para evaluar la biterapia con Herceptin y docetaxel tras la quimioterapia con AC o en combinación con docetaxel y carboplatino en pacientes con carcinoma de mama precoz HER2-positivo tras la cirugía. En el estudio HERA, el carcinoma de mama precoz se limitó a adenocarcinoma invasivo primario resecable de mama, con nódulos axilares positivos o nódulos axilares negativos si el tumor era de 1 cm de diámetro como mínimo. En el análisis conjunto de los estudios NCCTG N9831 y NSAPB B31, el cáncer de mama precoz se limitó a mujeres con cáncer de mama resecable de alto riesgo, definido como HER2-positivo y negativo para nódulos linfáticos axilares o HER2- positivo y negativo para nódulos linfáticos con características de alto riesgo (tamaño de tumor > 1 cm y ER-negativo o tamaño del tumor > 2 cm, independientemente del estado hormonal). En el estudio BCIRG 006, el carcinoma de mama precoz HER2-positivo se limitó a pacientes con ganglios linfáticos positivos o pacientes con ganglios negativos de alto riesgo, definidos como afectación ganglionar negativa (pN0), y al menos 1 de los factores siguientes: tamaño del tumor > 2 cm, ER-negativo y PR-negativo, grado histológico y/o nuclear 2-3 o edad < 35 años. En el uso neoadyuvante, se diseñó el estudio MO16432, multicéntrico y aleatorizado, para evaluar la utilidad clínica de la administración concomitante de Herceptin y quimioterapia neoadyuvante con una antraciclina y un taxano (AP+H seguido de P+H, seguido de CMF+H, seguido de Herceptin adyuvante, hasta una duración total del tratamiento de 1 año). Para este estudio se reclutaron pacientes con cáncer de mama recién diagnosticado localmente avanzado (estadio III) o inflamatorio. A las pacientes, con tumores HER2-positivos, se las aleatorizó para recibir, bien la quimioterapia neoadyuvante concomitante con Herceptin neoadyuvante/adyuvante, bien quimioterapia neoadyuvante sola. Los resultados del estudio HERA en cuanto a la eficacia se resumen en la tabla siguiente: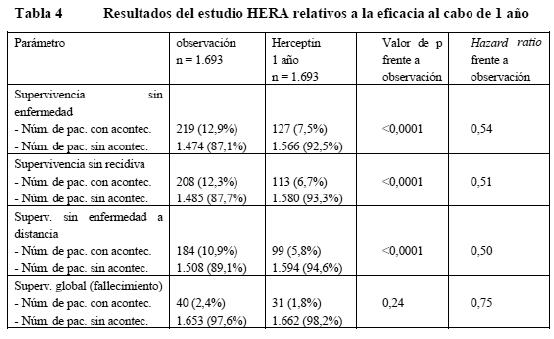
En la variable de valoración principal -la supervivencia sin enfermedad-, la hazard ratio (HR, razón de riesgos instantáneos) muestra un beneficio absoluto en cuanto a supervivencia sin enfermedad a los 2 años de 7,6 puntos porcentuales (85,8% frente al 78,2%) a favor del grupo con Herceptin. En los estudios NCCTG N9831 Y NSAPB B31, Herceptin se administró en combinación con paclitaxel, seguido de quimioterapia AC. Administración de paclitaxel: Paclitaxel i.v.: 80 mg/m2 en infusión i.v. continua cada semana, durante 12 semanas; Paclitaxel i.v: 175 mg/m2 en infusión i.v continua cada 3 semanas, durante 4 ciclos (día 1 de cada ciclo). En la tabla siguiente se resumen los resultados sobre la eficacia del análisis conjunto de los estudios NCCTG 9831 y NSABP B-31: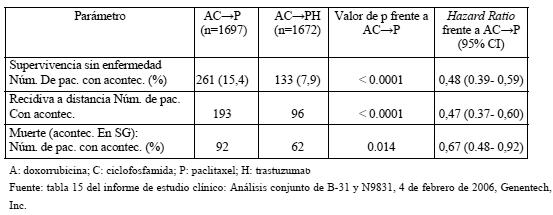
Por lo que respecta a la variable principal de valoración, la SSE, la adición de Herceptin paclitaxel se tradujo en un descenso del riesgo de recidiva del 52% (HR: 0,48 [IC del 95%: 0,39-0,59]; p < 0,0001). La HR muestra un beneficio absoluto en cuanto a supervivencia sin enfermedad a los 3 años de 11,8 puntos porcentuales (87,2% frente al 75,4%) a favor del grupo de AC- > PH (Herceptin). Resultados del primer análisis intermedio de la eficacia prospectivamente planificado: en la variable secundaria de valoración, la supervivencia global, el análisis conjunto de la eficacia de los estudios NSABP B-31 y NCCTG N9831 reveló una mejora estadísticamente significativa de la SG en los pacientes tratados con trastuzumab. La adición de trastuzumab a A- > CP redujo el riesgo de muerte en un 33%. La HR fue de 0,67 (IC del 95%: 0,48-0,92); p = 0,014. En el análisis conjunto de esta población, 154 pacientes aleatorizados habían fallecido; 92 pacientes (5,5%) en el grupo de AC- > P frente a 62 (3,7%) en el grupo AC- > PH. La mediana de seguimiento en el momento del análisis intermedio era de 1,8 años en el grupo de AC-+P y de 2,0 años en el de AC- > PH. En el estudio BCIRG 006, Herceptin se administró bien en combinación con docetaxel, seguido de quimioterapia AC (AC-DH), bien en combinación con docetaxel y carboplatino (DCarbH). Administración de docetaxel: docetaxel i.v.: 100 mg/m2 en infusión i.v. de 1 hora cada 3 semanas, durante 4 ciclos (día 2 del primer ciclo de docetaxel; después, día 1 de cada cielo siguiente). O Docetaxel i.v.: 75 mg/m2 en infusión i.v. de 1 hora cada 3 semanas, durante 6 ciclos (día 2 del primer ciclo de docetaxel; después, día 1 de cada ciclo). Y a continuación: Carboplatino: administración para obtener un ABC (área bajo la curva de concentraciones plasmáticas) diana de 6 mg/ml/min en infusión i.v. de 30-60 min cada 3 semanas, durante un total de 6 ciclos. En las tablas siguientes se resumen los resultados sobre la eficacia en el estudio BCIRG 006: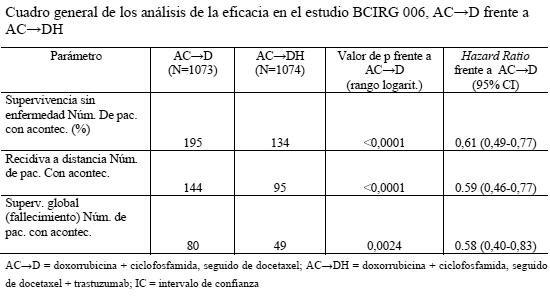
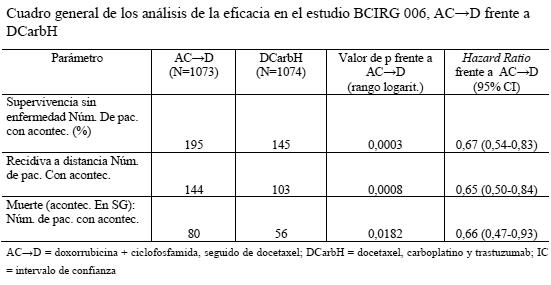
En el estudio BCIRG 006, por lo que respecta a la variable principal de valoración, la supervivencia sin enfermedad, la HR muestra un beneficio absoluto en cuanto a tasa de SSE a los 3 años de 5,8 puntos porcentuales (86,7 % frente a 80,9 %) a favor del grupo de AC→PH (Herceptin) y de 4,6 puntos porcentuales (85,5 % frente a 80,9 %) a favor del grupo de DCarbH (Herceptin) en comparación con AC→D. Por lo que respecta a la variable secundaria de valoración, la supervivencia global, la administración AC→DH redujo el riesgo de muerte en un 42% en comparación con AC→D (HR: 0,58 [IC del 95%: 0,40-0,83] p = 0,0024, prueba de rangos logarítmicos), y el riesgo de muerte disminuyó en un 34% en los pacientes tratados con DCarbH en comparación con los que recibieron AC→P (HR: 0,66 [IC del 95%: 0,47-0,93], p = 0,0182). En el segundo análisis intermedio del estudio BCIRG 006, 185 pacientes aleatorizados habían fallecido: 80 pacientes (7,5%) del grupo AC→D, 49 pacientes (4,6%) del grupo AC →DH y 56 pacientes (5,2%) del grupo DCarbH. La mediana de seguimiento fue de 2,9 años en el grupo AC→D y de 3,0 años en los grupos AC→DH y DCarbH. En el estudio MO16432 se administró Herceptin concomitantemente con 10 ciclos de quimioterapia neoadyuvante. La terapia se administró del modo siguiente: Doxorrubicina 60 mg/m2 y paclitaxel 150 mg/m2 y Herceptin (8 mg/kg de dosis inicial seguida 6 mg/kg de dosis de mantenimiento), administrados cada 3 semanas durante 3 ciclos; a continuación paclitaxel 175 mg/m2 y Herceptin 6 mg/kg, administrados cada 3 semanas durante ciclos; a continuación CMF los días 1 y 8 cada 4 semanas durante 3 ciclos, más 4 ciclos de Herceptin; a continuación hasta 7 ciclos adicionales de Herceptin solo (hasta completar 1 año desde el inicio de la administración de Herceptin). En la tabla siguiente se resumen los resultados relativos a la eficacia del estudio M016432. La mediana de la duración del seguimiento en el grupo de Herceptin fue de 3,8 años.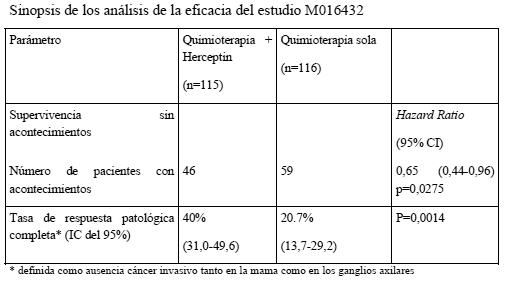
Por lo que se refiere a la variable principal de valoración, la supervivencia sin acontecimientos, la adición de Herceptin a la quimioterapia neoadyuvante seguida de Herceptin adyuvante durante un total de 52 semanas se tradujo en una reducción del 35% del riesgo de recurrencia/progresión de la enfermedad. La hazard ratio muestra un beneficio absoluto en cuanto a supervivencia sin acontecimientos a los 3 años de 13 puntos porcentuales (65 % frente al 52 %) a favor del grupo de Herceptin. En la Tabla 5 se resumen los resultados del estudio ToGA sobre la eficacia. Para este estudio se reclutaron pacientes no tratados previamente con adenocarcinoma HER2- positivo irresecable, localmente avanzado o recidivante y/o metastásico, de estómago o de la unión gastroesofágica no susceptible de tratamiento curativo. La variable principal de valoración era la supervivencia global, definida como el tiempo transcurrido entre la aleatorización y el fallecimiento por cualquier causa. En el momento del análisis habían fallecido en total 349 pacientes aleatorizados: 182 (62,8%) del grupo de control y 167 (56,8%) del grupo de tratamiento. La mayoría de las muertes se debieron a causas relacionadas con el cáncer subyacente. La supervivencia global mejoró significativamente en el grupo de Herceptin + capecitabina/5FU y cisplatino en comparación con el grupo de capecitabina/5-FU y cisplatino (p = 0,0046, prueba de rangos logarítmicos). La mediana de supervivencia fue de 11,1 meses con capecitabina/5-FU y cisplatino y de 13,8 meses con Herceptin + capecitabina/5-FU y cisplatino. El riesgo de fallecimiento disminuyó en un 26% (HR: 0,74, IC del 95%: 0,60-0,91) en el grupo de Herceptin en comparación con el de capecitabina/5FU. Análisis post-hoc de los subgrupos muestran que la acción selectiva sobre tumores con cifras altas de proteína HER2 (IHC 2+/FISH+ y IHC 3+/independientemente del resultado en una prueba FISH) se traduce en un mayor efecto terapéutico. La mediana de supervivencia global en el grupo con expresión alta de HER2 fue de 11,8 meses frente a 16 meses (HR: 0,65, IC del 95%: 0,51-0,83), y la mediana de supervivencia sin progresión, de 5,5 meses frente a 7,6 meses (HR: 0,64, IC del 95%: 0,51-0,79) en los grupos de capecitabina/5-FU y cisplatino y Herceptin + capecitabina/5-FU y cisplatino, respectivamente. En un estudio comparativo de métodos analíticos se halló un alto grado de concordancia ( > 95%) con las técnicas de SISH y FISH para detectar la amplificación del gen de HER2 en pacientes con cáncer gástrico.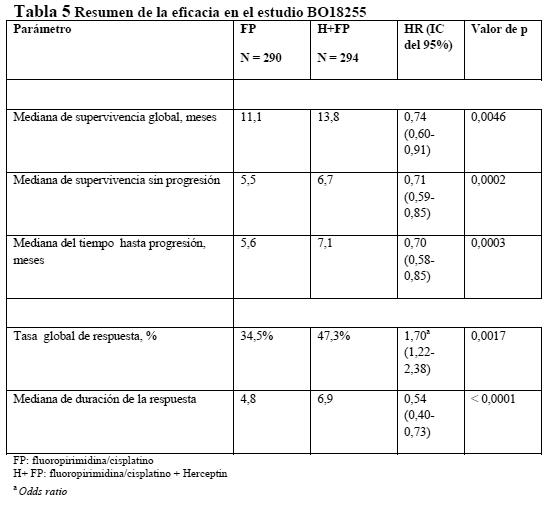
Inmunogenicidad: En 1 de 903 pacientes se detectaron anticuerpos humanos antitrastuzumab, sin que tuviera reacciones alérgicas. Propiedades farmacocinéticas: La farmacocinética del trastuzumab se ha estudiado en pacientes con carcinoma de mama metastásico, carcinoma de mama precoz y mediante un análisis poblacional de farmacocinética de pacientes con carcinoma gástrico avanzado. Con Herceptin no se han realizado estudios formales de interacción farmacológica. Carcinoma de mama: Infusiones intravenosas de corta duración de 10, 50, 100, 250 y 500 mg de trastuzumab una vez por semana pusieron de manifiesto una farmacocinética no lineal: el aclaramiento disminuía a medida que aumentaban las dosis. Farmacocinética en estado de equilibrio: Para estimar la farmacocinética en equilibrio en pacientes con carcinoma de mama metastásico tratadas con trastuzumab en una dosis de carga de 4 mg/kg seguida de una dosis de mantenimiento de 2 mg/kg/semana, se aplicó un método de farmacocinética poblacional utilizando datos obtenidos en estudios de fase I, fase II y estudios fundamentales de fase III. En esta evaluación, el aclaramiento típico de trastuzumab fue de 0,241 l/día (con un peso corporal de 68 kg); el volumen de distribución típico del compartimiento central VC, de 3,02 litros, y la semivida de eliminación correspondiente, de unos 28 días. Estos datos están respaldados por el informe más reciente de farmacocinética poblacional, en el que se describe un aclaramiento de 0,231 l/día con un volumen de distribución en los compartimientos central (Vc) y periférico (Vp) de 3,02 litros y 2,68 litros, respectivamente, en pacientes típicos, y la correspondiente semivida de eliminación de aproximadamente 38 días. Según todos los análisis poblacionales de farmacocinética, la semivida del trastuzumab es de 28-38 días, lo cual indica que la farmacocinética en equilibrio estacionario debe alcanzarse al cabo de unas 27 semanas (190 días o 5 semividas de eliminación). El mismo intervalo de tiempo correspondería a la eliminación del trastuzumab tras la suspensión del tratamiento con Herceptin. En los pacientes con carcinoma de mama precoz tratadas con Herceptin en una dosis de carga de 8 mg/kg, seguida de una dosis de mantenimiento de 6 mg/kg cada tres semanas, se alcanzaron concentraciones mínimas en equilibrio de 63 mg/l en el ciclo 13. Estas concentraciones eran comparables a las notificadas anteriormente en pacientes con carcinoma de mama metastásico. La quimioterapia concomitante (con antraciclinas/ciclofosfamida, paclitaxel o docetaxel) no parecía influir en la farmacocinética del trastuzumab. La administración concomitante de anastrozol no parecía influir en la farmacocinética del trastuzumab. Carcinoma gástrico avanzado: Farmacocinética en equilibrio estacionario en el carcinoma gástrico avanzado: Infusiones intravenosas de corta duración de 8 mg/kg de trastuzumab seguidos de 6 mg/kg cada 3 semanas en pacientes con carcinoma gástrico avanzado pusieron de manifiesto un aclaramiento dependiente de la concentración, compuesto principalmente de componentes lineales y no lineales con concentraciones séricas altas ( > 75 mg/ml) y bajas ( < 25 mg/ml), respectivamente. Para estimar la farmacocinética en equilibrio estacionario en pacientes con carcinoma gástrico avanzado tratados con trastuzumab cada 3 semanas en una dosis inicial de 8 mg/kg seguida de una dosis de mantenimiento de 6 mg/kg/semana cada 3 semanas, se aplicó un método de farmacocinética poblacional bicompartimental utilizando los datos del estudio de fase III B018255. En esta evaluación, el aclaramiento total está dominado por el aclaramiento lineal, y la semivida en los pacientes con carcinoma gástrico avanzado es de unos 26 días. La mediana del ABC previsto en equilibrio estacionario (a lo largo de un período de 3 semanas en equilibrio estacionario) es de 1.213mg•dia/l; la mediana de la Cmáx en equilibrio estacionario, de 128 mg/L, y la mediana de la Cmín en equilibrio estacionario, de 27,6 mg/l. No hay datos sobre la concentración de dominio extracelular circulante del receptor HER2 (shed antigen o antígeno circulante) en el suero de los pacientes con carcinoma gástrico. Farmacocinética en poblaciones especiales: No se han efectuado estudios farmacocinéticos específicos en pacientes ancianos o con insuficiencia renal o hepática. Ancianos: Se ha demostrado que la edad no altera la disposición farmacocinética del trastuzumab (ver Dosificación). Datos preclínicos sobre seguridad: Teratogenicidad: Se han llevado a cabo estudios en la reproducción de macacos de Java con dosis hasta 25 veces superiores a la dosis semanal de mantenimiento en el ser humano, de 2 mg/kg de Herceptin, sin que se apreciaran signos de fecundidad alterada o daño fetal. Sin embargo, al estimar el riesgo de toxicidad en la reproducción del ser humano, también es importante considerar la importancia de la forma del receptor HER2 en los roedores durante el desarrollo embrionario normal y la muerte embrionaria en ratones mutados sin este receptor. Se observó que el trastuzumab atravesaba la placenta durante el período de desarrollo fetal temprano (días 20-50 de la gestación) y tardío (días 120-150 de la gestación). Otros efectos: Lactancia: En un estudio realizado en macacos de Java lactantes con dosis de Herceptin 25 veces superiores a la dosis semanal de mantenimiento en el ser humano, de 2 mg/kg, se puso de manifiesto que el trastuzumab pasa a la leche materna. La presencia de trastuzumab en el suero de los macacos lactantes no se asoció a ningún efecto nocivo en el crecimiento o desarrollo de los mismos desde el nacimiento hasta el primer mes de edad.
Indicaciones.
Carcinoma de mama: Carcinoma de mama metastático (CMM): Herceptin está indicado para el tratamiento de pacientes con carcinoma de mama metastásico con sobreexpresión de HER2: a) como monoterapia en los pacientes que han recibido una o más pautas quimioterápicas previas contra su enfermedad metastásica; b) en politerapia con paclitaxel o docetaxel en los pacientes sin quimioterapia previa contra su enfermedad metastásica; c) en politerapia con un inhibidor de la aromatasa en los pacientes que presenten carcinoma de mama metastásico con positividad de los receptores hormonales. Carcinoma de mama precoz (CMP): Herceptin está indicado para el tratamiento del carcinoma de mama precoz HER2- positivo: tras la cirugía, la quimioterapia (neoadyuvante o adyuvante) y la radioterapia (si procede); tras la quimioterapia adyuvante con doxorrubicina y ciclofosfamida, en politerapia con paclitaxel o docetaxel; en combinación con quimioterapia adyuvante consistente en docetaxel y carboplatino. En combinación con quimioterapia neoadyuvante seguida de Herceptin adyuvante en el cáncer de mama localmente avanzado (incluido cáncer inflamatorio) o tumores > 2 cm de diámetro. Carcinoma gástrico avanzado: Herceptin en politerapia con capecitabina o 5-fluorouracilo i.v. y un derivado del platino está indicado para el tratamiento de paciente con adenocarcinoma gástrico o de la unión gastroesofágica HER2-positivo avanzado que no hayan recibido previamente tratamiento antineoplásico de su enfermedad metastásica.
Dosificación.
Antes de iniciar el tratamiento con Herceptin es obligatorio realizar una prueba de HER2. Herceptin debe administrarse en infusión i.v. Herceptin no debe administrarse en inyección i.v. rápida o bolo i.v. Pauta semanal: Dosis inicial: La dosis inicial (de carga) recomendada de Herceptin es de 4 mg/kg, en infusión i.v. de 90 minutos. Debe observarse si los pacientes presentan fiebre, escalofríos u otros síntomas relacionados con la infusión (ver Reacciones adversas). La interrupción de la infusión puede facilitar el control de estos síntomas. Tras su remisión, puede reanudarse la infusión. Dosis siguientes: La dosis semanal recomendada de Herceptin es de 2 mg/kg. Si la dosis anterior se toleró bien, puede administrarse la siguiente en infusión de 30 minutos. Debe observarse si los pacientes presentan fiebre, escalofríos u otros síntomas relacionados con la infusión (ver Reacciones adversas). Pauta alternativa cada tres semanas: Dosis inicial de 8 mg/kg, seguida de 6 mg/kg al cabo de 3 semanas; a continuación, 6 mg/kg cada 3 semanas, en infusión de aproximadamente 90 minutos. Si la dosis anterior se toleró bien, puede administrarse la siguiente en infusión de 30 minutos. Duración del tratamiento: En los estudios clínicos, pacientes con carcinoma de mama metastásico o carcinoma gástrico avanzado recibieron Herceptin hasta la progresión de la enfermedad. Los pacientes con carcinoma de mama precoz deben recibir tratamiento durante 1 año o hasta la recidiva de la enfermedad. Dosis no administradas: Si un paciente se salta una dosis de Herceptin en una semana o menos, se le debe administrar lo antes posible la dosis de mantenimiento habitual (pauta semanal: 2 mg/kg; pauta cada tres semanas: 6 mg/kg). No se debe esperar hasta el siguiente ciclo programado. Las dosis de mantenimiento siguientes de Herceptin (pauta semanal: 2 mg/kg; pauta cada tres semanas: 6 mg/kg) deben administrarse según el plan anterior. Si un paciente se salta una dosis de Herceptin en más de una semana, debe recibir una nueva dosis inicial en aproximadamente 90 minutos (pauta semanal: 4 mg/kg; pauta cada tres semanas: 8 mg/kg). Las dosis de mantenimiento siguientes de Herceptin (pauta semanal: 2 mg/kg; pauta cada tres semanas: 6 mg/kg) deben administrarse según el plan anterior. Reducción de las dosis: Durante los estudios clínicos no se efectuó ninguna reducción posológica de Herceptin. Los pacientes pueden continuar el tratamiento con Herceptin durante los períodos de mielodepresión reversible inducida por la quimioterapia, pero se los debe vigilar estrechamente para detectar posibles complicaciones de una neutropenia durante esos períodos. Se observarán las instrucciones específicas de reducción o mantenimiento posológico de la quimioterapia. Pautas posológicas especiales: Ancianos: Los datos sugieren que la edad no altera la disposición farmacocinética de Herceptin (ver Farmacocinética en poblaciones especiales). En los ensayos clínicos, los pacientes ancianas no recibieron dosis reducidas de Herceptin. Niños: No se han estudiado la seguridad y la eficacia de Herceptin en pacientes de edad infantil.
Contraindicaciones.
Herceptin está contraindicado en pacientes alérgicos al trastuzumab o a cualquier otro componente del producto.
Reacciones adversas.
Ensayos clínicos: En este apartado se utilizan las siguientes categorías de frecuencia de las reacciones adversas: muy frecuente (≥1/10), frecuente (≥1/100 a < 1/10), poco frecuente ( > 1/1.000 a < 1/100), rara vez ( > 1/10.000 a < 1/1.000), muy rara vez ( < 1/10.000), desconocida (no estimable a partir de los datos disponibles). Dentro de cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad. Lista de reacciones adversas: En la tabla siguiente se presentan las reacciones adversas notificadas en asociación con el uso de Herceptin solo o en combinación con quimioterapia en estudios clínicos fundamentales. De todos los términos incluidos se indica el porcentaje más alto registrado en los estudios clínicos fundamentales. Dado que Herceptin se utiliza habitualmente con otros quimioterápicos y radioterapia, a menudo es difícil establecer la relación causal de un efecto secundario con un fármaco determinado o la radioterapia.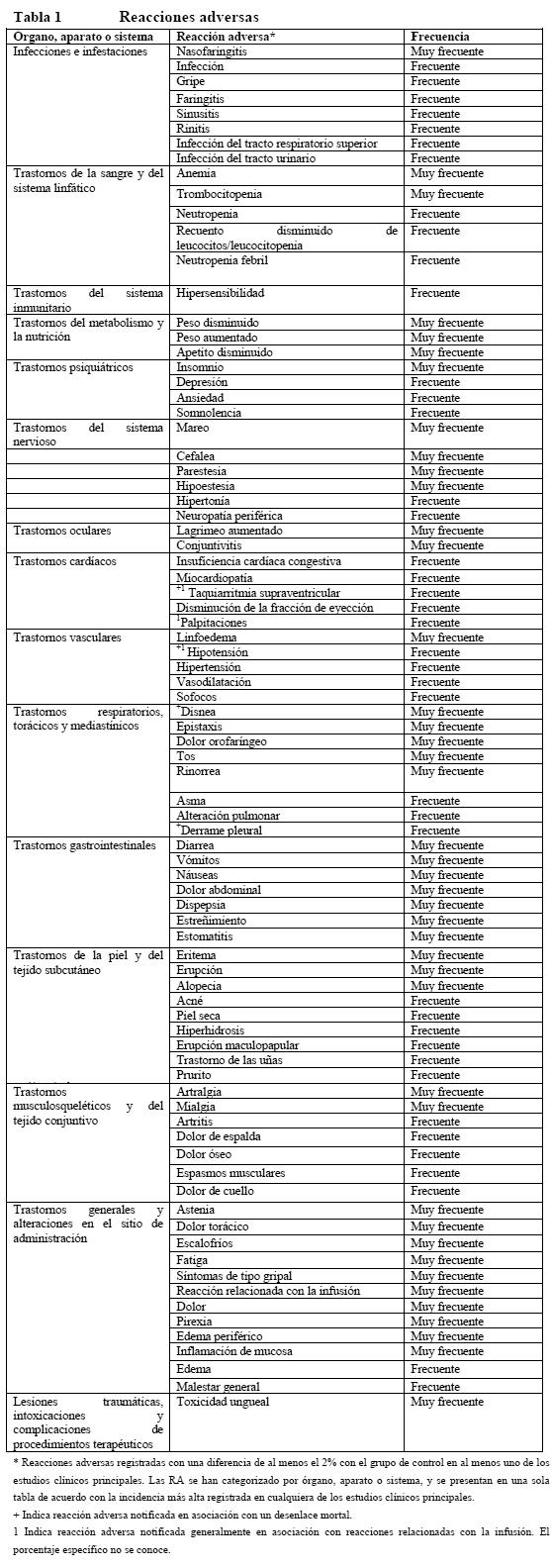
La información siguiente es de interés para todas las indicaciones: Síntomas relacionados con la infusión: Durante la primera infusión de Herceptin suelen observarse escalofríos o fiebre o ambas cosas. Otros signos y síntomas posibles son: náuseas, vómitos, dolor, escalofríos, cefalea, tos, mareos, exantema, astenia e hipertensión. Por lo general, estos síntomas son leves o moderados y no suelen presentarse con las infusiones de Herceptin ulteriores. Como tratamiento de estos síntomas pueden administrarse analgésicos o antipiréticos, como meperidina y paracetamol, o antihistamínicos, como difenhidramina (ver Dosificación). Algunas reacciones adversas a la infusión de Herceptin, como disnea, hipotensión, sibilancias, broncospasmo, taquicardia, saturación de oxígeno disminuida y dificultad respiratoria, pueden ser graves e incluso tener una evolución fatal (ver Advertencias). Reacciones de hipersensibilidad: En casos aislados se han observado reacciones anafilactoides: Cardiotoxicidad: Carcinoma de mama: En los pacientes tratadas con Herceptin se han observado signos y síntomas de insuficiencia cardíaca como disnea, ortopnea, tos elevada, edema pulmonar, ritmo de galope (S3) o disminución de la fracción de eyección (ver Advertencias y precauciones). Según los criterios aplicados para definir la disfunción cardíaca, la incidencia en los estudios clínicos fundamentales con enfermedad metastásica osciló entre el 9% y el 12% en el subgrupo de Herceptin y paclitaxel, frente al 1-4 % en el grupo tratado con paclitaxel en monoterapia. Con Herceptin en monoterapia, la tasa fue del 6- 9%. En los pacientes tratados con Herceptin y antraciclina/ciclofosfamida (27-28%) se registró la tasa más alta de disfunción cardíaca, la cual fue significativamente superior a la notificada en los pacientes que sólo recibieron antraciclina/ciclosfosfamida (7-10%). En un estudio posterior con control prospectivo de la función cardíaca, la incidencia de insuficiencia cardíaca sintomática fue del 2,2% en los pacientes tratados con Herceptin y docetaxel, frente al 0% en las que recibieron docetaxel en monoterapia. En el estudio HERA se observó insuficiencia cardíaca de clases III-IV de la NYHA en el 0,6% de los pacientes del grupo de un año. Dado que la semivida terminal media de Herceptin es de aproximadamente 3 semanas, trastuzumab puede permanecer en la circulación hasta 15 semanas después de retirado el tratamiento. La utilización de una antraciclina en este período podría comportar un mayor riesgo de disfunción cardíaca; por ello, se recomienda sopesar con detenimiento los riesgos y los beneficios esperados, además de vigilar estrechamente la función cardíaca. Carcinoma gástrico avanzado: En el estudio ToGA, la mediana de FEVI en el cribado fue del 64% (extremos: 48% y 90%) en el grupo de fluoropirimidina /cisplatino (FP),y del 65% (extremos: 50% y 86%) en el grupo de Herceptin y fluoropirimidina /cisplatino (H + FP). En la mayoría de los casos, la disminución de la FEVI observada en el estudio ToGA fue asintomática, excepción hecha de un paciente del grupo de Herceptin cuyo descenso de la FEVI coincidió con insuficiencia cardíaca. Resumen de los cambios de la FEVI desde el cribado: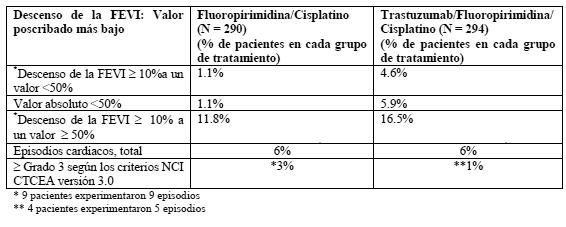
Acontecimientos adversos cardiacos:
En conjunto, no hubo diferencias significativas de cardiotoxicidad entre el grupo de tratamiento y el grupo comparativo. Toxicidad hematológica: Carcinoma de mama: La toxicidad hematológica es infrecuente tras la administración de Herceptin en monoterapia a pacientes con enfermedad metastásica. Se han producido leucocitopenia, trombocitopenia y anemia de grado 3 de la OMS en < 1% de los pacientes. No se han observado reacciones adversas de grado IV de la OMS. En los pacientes tratados con Herceptin y paclitaxel se registró un incremento de la toxicidad hematológica de grados III o IV de la OMS en comparación con los que recibieron paclitaxel solo (34% y 21 %, respectivamente). La toxicidad hematológica también aumentó en los pacientes que recibieron Herceptin y docetaxel en comparación con los tratados con docetaxel solo (32% frente al 22% con neutropenia de grados 3-4 según los criterios NCI-CTC). La incidencia de neutropenia febril/sepsis neutropénica también creció en los pacientes tratados con Herceptin y docetaxel (23% frente al 17% en los que recibieron docetaxel en monoterapia). De acuerdo con los criterios NCI-CTC, en el estudio HERA hubo un 0,4% de pacientes tratadas con Herceptin que experimentaron un cambio de 3 o 4 grados del valor basal, frente al 0,6% en el grupo de observación. Carcinoma gástrico avanzado: A continuación se muestran los AA más frecuentes de grado 3 o superior con una incidencia de al menos un 1% por tratamiento ensayado en la categoría de trastornos de la sangre y del sistema linfático (clasificación por órganos y sistemas, SOC):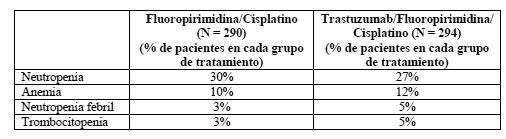
EI porcentaje total de pacientes con AA de grado 3 o superior según los criterios NCI-CTCAE v3.0 perteneciente a esta categoría fue del 38% en el grupo de FP y el 40% en el grupo de FP+H. En conjunto, no hubo diferencias significativas de hematotoxicidad entre el grupo de tratamiento y el grupo comparativo. Toxicidad hepática y renal: Carcinoma de mama: Se ha observado toxicidad hepática de grado 3 o 4 de la OMS en el 12% de los pacientes tras la administración de Herceptin en monoterapia contra la enfermedad metastásica. Esta toxicidad se ha asociado con progresión de la enfermedad en el hígado en el 60% de estos pacientes. La toxicidad hepática de grado 3 o 4 de la OMS se observó con menor frecuencia en los pacientes tratadas con Herceptin y paclitaxel que entre los pacientes tratadas con paclitaxel solo (7% y 15%, respectivamente). No se observó toxicidad renal de grado 3 o 4 de la OMS. Carcinoma gástrico avanzado: En el estudio ToGA no se registraron diferencias significativas de toxicidad hepática y renal entre los dos grupos de tratamiento. La toxicidad renal de grado 3 o superior según los criterios NCI-CTCAE no fue significativamente mayor en los pacientes tratados con Herceptin que en los del grupo de FP (3% y 2%, respectivamente). Acontecimientos adversos de grado 3 o superior según los criterios NCI-CTCAE (v. 3.0) en la categoría trastornos hepatobiliares de la clasificación SOC: hiperbilirrubinemia fue el único AA notificado y no fue significativamente mayor en los pacientes tratados con Herceptin que en los del grupo de FP (1% y < 1%, respectivamente). Diarrea: El 27% de los pacientes tratados con Herceptin en monoterapia contra la enfermedad metastásica experimentaron diarrea. También se ha observado un aumento de la incidencia de diarrea, sobre todo de intensidad leve o moderada, entre los pacientes tratados con Herceptin y paclitaxel, en comparación con los que recibieron paclitaxel en monoterapia. En el estudio HERA, el 7% de los pacientes tratados con Herceptin experimentaron diarrea. Carcinoma gástrico avanzado: En el estudio ToGA sufrieron diarrea de cualquier grado 109 pacientes (37%) del grupo de Herceptin frente a 80 (28%) del grupo comparativo. Según los criterios NCI-CTCAE v. 3.0, el porcentaje de pacientes con diarrea de grado 3 o superior fue del 4% en el grupo de FP frente al 9% en el grupo de FP+H. Infecciones: Se ha registrado una mayor incidencia de infecciones, principalmente de infecciones leves de las vías respiratorias altas de poca importancia clínica o de infecciones por catéter, en los pacientes tratados con Herceptin. Alteraciones analíticas: Neutropenia febril se produce muy frecuentemente. Reacciones adversas frecuentes son la anemia, la leucocitopenia, la trombocitopenia y la neutropenia. La frecuencia de hipoprotrombinemia no se conoce. Experiencia tras la comercialización: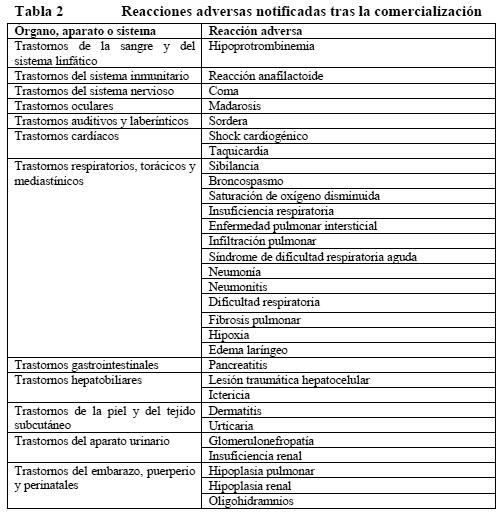
Acontecimientos adversos: La Tabla 3 recoge los acontecimientos adversos históricamente notificados en pacientes tratados con Herceptin. No se ha hallado ninguna relación causal entre Herceptin y estos acontecimientos adversos, por lo cual no se considera que estén previstos con fines de notificación reglamentaria.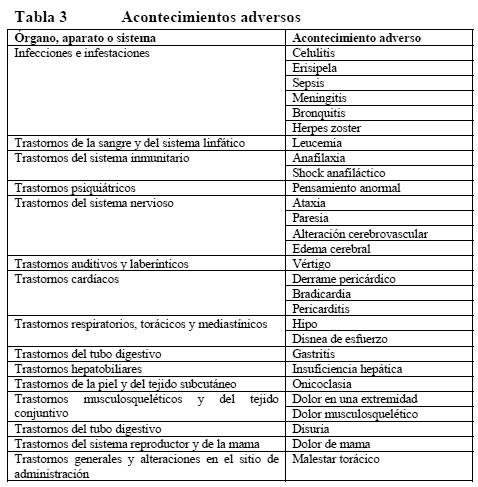
Advertencias.
El tratamiento con Herceptin debe iniciarse únicamente bajo la supervisión de un médico especializado en el tratamiento de pacientes oncológicos. Reacciones adversas graves descritas con poca frecuencia tras la infusión de Herceptin han sido: disnea, hipotensión, sibilancias, broncospasmo, taquicardia, saturación de oxígeno reducida y dificultad respiratoria. De observarse alguno de estos síntomas, debe suspenderse la infusión de Herceptin hasta su resolución. Con medidas de apoyo como la administración de oxígeno, beta-agonistas o corticosteroides se han tratado con éxito las reacciones graves (ver Reacciones adversas). En raras ocasiones, estas reacciones han tenido una evolución clínica con desenlace fatal. Los pacientes con disnea de reposo a causa de complicaciones neoplásicas o de comorbilidad pueden correr un riesgo mayor de sufrir una reacción fatal a la infusión. Dado que el tratamiento de estas pacientes exige una precaución extrema, conviene sopesar sus riesgos y beneficios individualmente (ver Reacciones adversas). Se han descrito efectos secundarios pulmonares graves tras la comercialización de Herceptin. Ocasionalmente, estos episodios adversos tuvieron un desenlace fatal. Se han notificado asimismo casos de enfermedad pulmonar intersticial, incluido infiltrado pulmonar, síndrome de dificultad respiratoria aguda, neumonía, neumonitis, derrame pleural, dificultad respiratoria, edema pulmonar agudo e insuficiencia respiratoria. Estos efectos pueden presentarse como parte de una reacción asociada a la infusión o con algún retardo. Los pacientes con neumopatía intrínseca sintomática o una extensa afectación tumoral de los pulmones, causante de disnea de reposo, pueden correr un riesgo mayor de reacciones graves (ver Reacciones adversas). Entre los factores de riesgo asociados con enfermedad pulmonar intersticial se hallan los tratamientos previos o concomitantes con otros antineoplásicos que se sabe están asociados con ella, como los taxanos, la gemcitabina, la vinorelbina y la radioterapia. Estos efectos pueden presentarse como parte de una reacción asociada a la infusión o con algún retardo. Los pacientes con disnea de reposo debida a complicaciones de una neoplasia avanzada y factores de riesgo asociados pueden correr un mayor riesgo de episodios pulmonares. Por consiguiente, tales pacientes no deben recibir Herceptin. Se ha observado insuficiencia cardíaca (clases II-IV de la clasificación de la New York Heart Association [NYHA]) en pacientes tratados con Herceptin en monoterapia o en asociación con paclitaxel después de un régimen quimioterápico que contenía antraciclinas (doxorrubicina o epirrubicina). Esta reacción puede ser moderada o grave, y se ha asociado con el fallecimiento de la paciente afectada (ver Reacciones adversas). Especial precaución exige el tratamiento de pacientes con insuficiencia cardíaca sintomática, antecedentes de hipertensión o coronariopatía documentada, así como, en el carcinoma de mama precoz, de pacientes con una fracción de eyección ventricular izquierda (FEVI) del 55% o menor. Los pacientes candidatos al tratamiento con Herceptin, sobre todo los que hayan recibido anteriormente antraciclinas y ciclofosfamida, deben someterse a un reconocimiento médico para evaluar el estado cardíaco que comprenda anamnesis, exploración física, ECG, ecocardiografía y/o ventriculografía isotópica (MUGA). De igual modo, antes de decidir si se administra Herceptin, debe realizarse un cuidadoso análisis de riesgos y beneficios. En el CMP se excluyó a los pacientes señaladas a continuación del estudio HERA; por consiguiente, no hay da
