HUMIRA®
ABBOTT
Inhib. del factor de necrosis tumoral.
Descripción.
Humira (adalimumab) es un anticuerpo monoclonal inmunoglobulínico (IgG1) recombinante humano que contiene únicamente secuencias peptídicas humanas. Humira se creó por medio de la técnica de presentación de fagos, que permite aislar regiones variables de cadenas pesadas y livianas de Ig totalmente humanas, que confieren especificidad al factor de necrosis tumoral (TNF) humano y las secuencias de cadena liviana kappa y cadena pesada de IgG1 humana. Humira se une con gran afinidad y especificidad al factor de necrosis tumoral soluble (TNF-alfa), pero no a la linfotoxina (TNF-beta). Adalimumab se obtiene mediante tecnología de ADN recombinante en un sistema de expresión de células de mamífero. Está constituido por 1.330 aminoácidos y tiene un peso molecular de aproximadamente 148 Kilodaltons. Humira se proporciona como una solución estéril, sin preservantes para administración subcutánea de adalimumab. El producto se proporciona como una jeringa de vidrio de uso único prellenada con 1 mL, o un frasco ampolla de vidrio de uso único con 1 mL, o un Pen (Humira Pen) prellenada de uso único. Dentro del pen está la jeringa de vidrio prellenada de uso único con 1 mL. La solución de Humira es transparente e incolora, con un pH de alrededor de 5,2. Cada jeringa de uso único prellenada de Humira contiene 40 mg de adalimumab por 0.8 mL (50 mg/mL). Los ingredientes inactivos incluyen: 4.93 mg de cloruro sódico, 0.69 mg de fosfato sódico monobásico dihidratado, 1.22 mg de fosfato sódico dibásico dihidratado, 0.24 mg de citrato sódico, 1.04 mg de ácido cítrico monohidratado, 9.6 mg de manitol, 0.8 mg de polisorbato 80 y agua para inyección para 0.8 mL.
Farmacología.
General: Adalimumab se une específicamente al TNF y neutraliza la función biológica del TNF al bloquear su interacción con los receptores de superficie celular p55 y p75 del TNF. El TNF es una citoquina natural que interviene en respuestas inflamatorias e inmunológicas normales. Se encuentran concentraciones elevadas de TNF en el líquido sinovial de pacientes con artritis reumatoidea, incluyendo artritis idiopática juvenil, artritis psoriática y Espondilitis Anquilosante y tiene un papel importante tanto en la inflamación patológica como en la destrucción articular, que son características de estas enfermedades. También se encontraron niveles elevados de TNF en las placas de psoriasis. En psoriasis en placa, el tratamiento con Humira puede disminuir el grosor epidérmico y la infiltración de células inflamatorias. La relación entre estas actividades farmacodinámicas y el(los) mecanismo(s) por el cual Humira ejerce sus efectos clínicos se desconoce. Adalimumab también modula las respuestas biológicas inducidas o reguladas por TNF, incluidos los cambios en las concentraciones de las moléculas de adhesión responsables de la migración de leucocitos (ELAM-1, VCAM-1 e ICAM-1 con una IC50 de 1-2 x 10-10 M). Farmacodinámica: Después del tratamiento con Humira, se observó una rápida disminución en los niveles de reactantes de fase aguda de la inflamación (proteína C reactiva [PCR] y velocidad de sedimentación globular [VSG]) y citoquinas séricas (IL-6) en comparación con los valores basales en pacientes con AR. También se observó una rápida disminución en los niveles de PCR en pacientes con AIJ o enfermedad de Crohn. Los niveles séricos de las metaloproteinasas de la matriz (MMP-1 y MMP-3) que producen la remodelación tisular responsable de la destrucción del cartílago, también disminuyeron tras la administración de Humira. Los pacientes con AR, APs y EA a menudo presentan una anemia leve a moderada y una disminución del recuento de linfocitos, así como un aumento del recuento de neutrófilos y plaquetas. Los pacientes tratados con Humira generalmente experimentaron una mejoría de estos signos hematológicos de inflamación crónica. La relación concentración sérica-eficacia de adalimumab conforme a los criterios de respuesta del American College of Rheumatology (ACR 20) parece seguir la ecuación de Emáx de Hill, como se muestra a continuación:
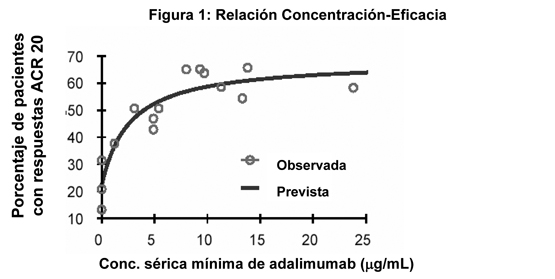
Se obtuvieron estimados de CE50 desde 0,8 a 1,4 mg/mL a través del modelo farmacocinético/farmacodinámico del recuento de articulaciones inflamadas, el recuento de articulaciones dolorosas y la respuesta ACR 20 de pacientes que participaron en estudios fases II y III. Farmacocinética: Absorción: Después de la administración de una dosis subcutánea (SC) única de 40 mg de Humira a 59 sujetos adultos sanos, la absorción y distribución de adalimumab fueron lentas, alcanzándose la concentración media sérica máxima aproximadamente cinco días después de la administración. La biodisponibilidad promedio absoluta de adalimumab estimada a partir de tres estudios después de una dosis subcutánea única de 40 mg, fue de 64%. Distribución y eliminación: Se determinó la farmacocinética de dosis única de adalimumab en varios estudios con dosis intravenosas de entre 0.25 y 10 mg/kg. El volumen de distribución (Vss) varió entre 4.7 y 6.0 L, lo que indica que adalimumab se distribuye de manera aproximadamente uniforme entre el líquido vascular y extravascular. Adalimumab se elimina lentamente, con clearances habituales por debajo de 12 mL/h. La media de la vida media de la fase terminal fue de aproximadamente dos semanas, en un rango de 10 a 20 días en los diferentes estudios. El clearance y la vida media no cambiaron relativamente a través del rango de dosis estudiado y la vida media terminal fue similar tras la administración intravenosa y subcutánea. Las concentraciones de adalimumab en el líquido sinovial de varios pacientes con AR variaron entre 31% a 96% con respecto a los valores en el suero. Farmacocinética en estado de equilibrio: La acumulación de adalimumab fue predecible basándose en la vida media tras la administración subcutánea de 40 mg de Humira semana por medio a pacientes con AR, con concentraciones medias mínimas en estado de equilibrio de aproximadamente 5 mcg/mL (sin metotrexato (MTX) concomitante) y 8 a 9 mcg/mL (con MTX concomitante), respectivamente. Las concentraciones séricas mínimas en estado de equilibrio de adalimumab aumentaron de manera aproximadamente proporcional con la dosis tras la administración de 20, 40 y 80 mg semana por medio y una vez a la semana por vía subcutánea. En estudios a largo plazo con un tratamiento de más de dos años no se observaron cambios en el clearance con el tiempo. En pacientes con psoriasis, la media de la concentración mínima estado estable fue 5 mcg/mL durante el tratamiento con monoterapia de adalimumab 40 mg semana por medio. Los análisis farmacocinéticos de población con datos de más de 1200 pacientes revelaron que la administración concomitante de MTX tuvo un efecto intrínseco sobre el clearance aparente de adalimumab (CL/F) (ver Interacciones). Como se esperaría, se observó una tendencia a un clearance aparentemente mayor de adalimumab con un aumento del peso corporal y en presencia de anticuerpos anti-adalimumab. También se identificaron otros factores menos importantes; se pronosticó un clearance aparentemente mayor en pacientes tratados con dosis menores a la dosis recomendada, así como en pacientes con concentraciones elevadas de factor reumatoideo o PCR. Estos factores probablemente no sean importantes desde el punto de vista clínico. En pacientes con enfermedad de Crohn, la dosis de carga de 160 mg de Humira en la Semana 0 seguida por 80 mg de Humira en la Semana 2 alcanza niveles medios séricos mínimos de adalimumab de aproximadamente 12 mcg/mL en la Semana 2 y Semana 4. Una media de niveles mínimos de estado estable de aproximadamente 7 mcg/mL se observó en la Semana 24 y Semana 56 en los pacientes con enfermedad de Crohn después de recibir una dosis de 40 mg de Humira semana por medio. Poblaciones especiales: Se investigó la farmacocinética en poblaciones especiales por medio de análisis farmacocinéticos de poblaciones. Ancianos: La edad pareció tener un efecto mínimo sobre el clearance aparente de adalimumab. En los análisis de poblaciones, los clearances medios ajustados por el peso en pacientes de 40 a 65 años de edad (n = 850) y ≥ 65 años (n = 287) fueron de 0.33 y 0.30 mL/h/kg, respectivamente. Niños: Después de la administración de 24 mg/m2 (hasta un máximo de 40 mg) subcutáneamente semana por medio a pacientes con artritis idiopática juvenil poliarticular (AIJ) la concentración sérica media mínima estado estable de adalimumab (valores medidos desde la Semana 20 a 48) fue 5.6 ± 5.6 mg/mL (102% CV) en Humira monoterapia y 10.9 ± 5.2 mg/mL (47.7% CV) con metotrexato concomitante. Las concentraciones séricas medias mínimas estado estable de adalimumab para sujetos que pesan < 30 kg que recibieron 20 mg Humira subcutáneamente semana por medio como monoterapia o con metrotrexato concomitante fueron 6.8 mg/mL y 10.9 mg/mL, respectivamente. Las concentraciones séricas medias mínimas estado estable para sujetos que pesan ≥ 30 kg que recibieron 40 mg Humira subcutáneamente semana por medio como monoterapia o con metrotrexato concomitante fueron 6.6 mg/mL y 8.1 mg/mL, respectivamente. Género: No se observaron diferencias farmacocinéticas relacionadas con el género después de realizar una corrección en función del peso corporal. Raza: No cabe esperar diferencias en el clearance de inmunoglobulinas entre razas. A partir de información limitada sobre individuos no caucásicos, no se observaron diferencias farmacocinéticas importantes para adalimumab. Insuficiencia hepática y renal: No se dispone de datos farmacocinéticos de pacientes con insuficiencia hepática o renal. Enfermedades: Los voluntarios sanos y los pacientes con AR mostraron una farmacocinética de adalimumab similar. Datos de seguridad pre-clínicos: Los datos preclínicos no indican un riesgo especial para el ser humano basándose en estudios de toxicidad de dosis única, toxicidad por dosis repetidas y genotoxicidad. Carcinogénesis, Mutagénesis y Alteración de la Fertilidad: No se han realizado estudios a largo plazo de adalimumab en animales para evaluar el potencial carcinogénico o su efecto sobre la fertilidad. No se observaron efectos clastogénicos ni mutagénicos de adalimumab en la prueba de micronúcleos en ratones in vivo ni en la prueba de Salmonella-Escherichia coli (Ames), respectivamente. Descripción de estudios clinicos: Estudios clínicos con artritis reumatoidea: Humira se evaluó en más de 3000 pacientes en todos los estudios clínicos de artritis reumatoidea. Algunos pacientes se trataron por más de 60 meses. La eficacia y la seguridad de Humira fueron evaluadas en cinco estudios randomizados, doble ciego y bien controlados. El estudio I en AR (DE009) evaluó 271 pacientes con AR moderada a severamente activa ≥ 18 años de edad, que no habían respondido al tratamiento con al menos uno pero no más de cuatro fármacos antirreumáticos modificadores de enfermedad (DMARDs) (p. Ej., hidroxicloroquina, oro oral o inyectable, azatioprina, D-penicilamina, sulfasalazina), y tuvieron una eficacia insuficiente con MTX en dosis de 12.5 a 25 mg (10 mg si eran intolerantes a MTX) por semana y en los que la dosis de MTX se mantuvo constante en 10 a 25 mg por semana. Los pacientes tenían ≥ 6 articulaciones inflamadas y ≥ 9 articulaciones dolorosas y una AR diagnosticada de acuerdo a los criterios ACR. Se administraron dosis de 20, 40 u 80 mg de Humira o placebo semana por medio durante 24 semanas. El estudio II en AR (DE011) evaluó 544 pacientes con AR moderada a severamente activa que tenían ≥ 18 años de edad y que no habían respondido al tratamiento con al menos un DMARD (por ej., MTX, sulfasalazina, hidroxicloroquina, oro oral o inyectable, D-penicilamina, azatioprina). Los pacientes tenían ≥ 10 articulaciones inflamadas y ≥ 12 articulaciones dolorosas y también fueron diagnosticados de acuerdo a los criterios ACR. Se administraron dosis de 20 ó 40 mg de Humira por inyección subcutánea semana por medio con placebo en las semanas alternas o cada semana durante 26 semanas; se administró placebo cada semana durante el mismo período de tiempo. El estudio III en AR (DE019) evaluó 619 pacientes con AR moderada a severamente activa que tenían ≥ 18 años de edad, tenían eficacia insuficiente a MTX en dosis de 12.5 a 25 mg (10 mg si eran intolerantes a MTX) cada semana y en los que la dosis de MTX se mantuvo constante en 12.5 a 25 mg cada semana. A diferencia del estudio I en AR, no era necesario que los pacientes en el estudio III en AR hubieran fallado al tratamiento con algún DMARDs con excepción de MTX. Los pacientes tenían ≥ 6 articulaciones inflamadas y ≥ 9 articulaciones dolorosas y se diagnosticó AR de acuerdo a los criterios ACR. En este estudio hubo tres grupos. El primero recibió inyecciones de placebo cada semana durante 52 semanas. El segundo recibió 20 mg de Humira cada semana durante 52 semanas. El tercer grupo recibió 40 mg de Humira semana por medio con inyecciones de placebo las otras semanas. Al final de las primeras 52 semanas, 457 pacientes fueron reclutados en una fase de extensión abierta en la cual recibieron 40 mg de Humira/MTX semana por medio hasta 60 meses. El estudio IV en AR (DE031) evaluó 636 pacientes con AR moderada a severamente activa que tenían ≥ 18 años de edad. Estos pacientes cumplían con los criterios ACR para diagnóstico de AR durante al menos tres meses y tenían al menos 6 articulaciones inflamadas y 9 articulaciones dolorosas. Se permitió que los pacientes fueran ya sea naive para DMARD o que mantuvieran su tratamiento reumatológico previo siempre que éste fuera estable durante un mínimo de 28 días. Se distribuyó aleatoriamente a los pacientes para recibir 40 mg de Humira o placebo semana por medio durante 24 semanas. El estudio V en AR (DE013) evaluó 799 pacientes adultos, naive para metotrexato, con artritis reumatoidea temprana moderada a severamente activa (duración media de la enfermedad menor de 9 meses). Este estudio evaluó la eficacia de la terapia combinada de Humira 40mg semana por medio/ metotrexato, monoterapia con Humira 40 mg semana por medio y monoterapia con metotrexato en la reducción de los signos y síntomas de AR y el índice de progresión del daño articular en artritis reumatoidea por 104 semanas. Los resultados de los cinco estudios se expresaron como porcentaje de pacientes con mejoría de la AR utilizando los criterios de respuesta ACR. El objetivo primario en los estudios I, II y III en AR y el objetivo secundario en el estudio IV en AR fue el porcentaje de pacientes que alcanzaron una respuesta ACR 20 en la semana 24 ó 26. El objetivo primario en el estudio V en AR fue el porcentaje de pacientes que alcanzaron una respuesta ACR 50 en la semana 52. Los estudio III y V en AR tenían un objetivo primario adicional a las 52 semanas de retraso en la progresión de la enfermedad (detectado en los resultados radiográficos). El estudio III en AR también tenía un objetivo primario de cambios en la calidad de vida. Respuesta clínica: Estudios I, II y III: El porcentaje de pacientes tratados con Humira que alcanzaron respuestas ACR 20, 50 y 70 fue consistente a través de los tres estudios. Los resultados de los tres estudios se resumen en la tabla 3.
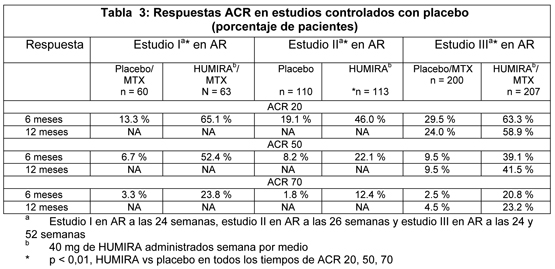
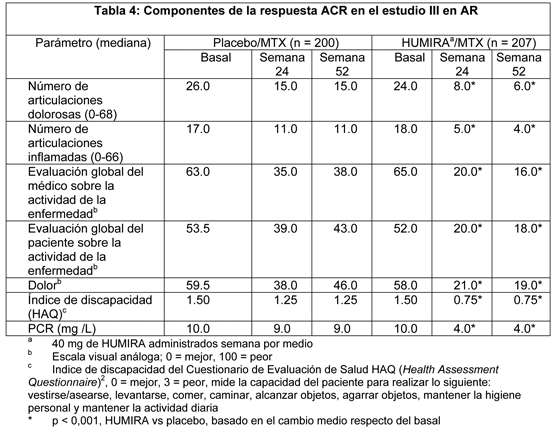
Los pacientes que recibieron Humira 40 mg cada semana en el estudio II en AR también alcanzaron tasas de respuesta ACR 20, 50 y 70 estadísticamente significativas de 53.4%, 35.0 % y 18.4 %, respectivamente, a los seis meses. En la tabla 4 se muestran los resultados de los componentes de los criterios de respuesta ACR del estudio III en AR. Los resultados presentados a continuación son generalmente representativos de cada estudio realizado. Las tasas de respuesta ACR y la mejoría en todos los criterios de respuesta ACR se mantuvieron a la semana 104. Después de 2 años en el estudio III en AR, el 24% de los pacientes con Humira alcanzaron una respuesta clínica importante, definida como mantención de una respuesta ACR 70 después de un período de 6 meses. Las respuestas ACR se mantuvieron en proporciones similares de los pacientes por hasta 60 meses con tratamiento continuo con Humira en la fase abierta del estudio III en AR.
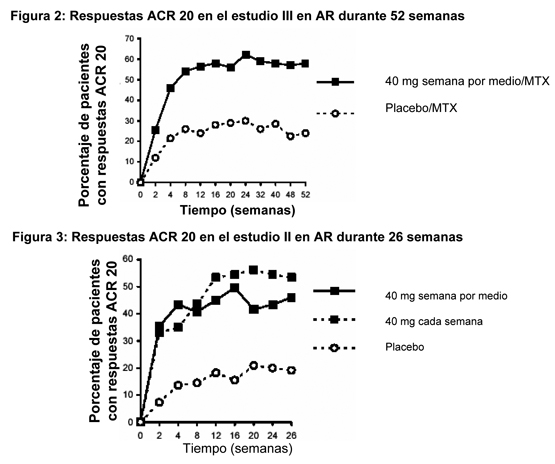
En el estudio III en AR, 84,7 % de los pacientes con respuestas ACR 20 en la semana 24 mantuvo la respuesta a las 52 semanas. Las siguientes figuras ilustran la duración de las respuestas ACR 20 a HUMIRA en los estudios III y II.
Estudio IV en AR: La respuesta ACR 20 de los pacientes tratados con Humira más cuidados estándar fue estadísticamente significativamente mejor que en pacientes tratados con placebo más cuidados estándar (p < 0,001). No se observaron reacciones adversas específicas relacionadas con la combinación de Humira y otros DMARDs. En los estudios I-IV en AR, los pacientes tratados con Humira alcanzaron respuestas ACR 20, 50 y 70 con mayor rapidez y con mayor frecuencia que los pacientes tratados con placebo. En el estudio I en AR, se observó una diferencia estadísticamente significativa en las respuestas ACR 20 en la semana 1 (primera visita del estudio) entre los pacientes tratados con Humira (26.0 %) y placebo (5.0 %). También se observaron diferencias estadísticamente significativas en las respuestas ACR 20 en los estudios II, III y IV en AR en la semana 2 (primera visita del estudio) entre los pacientes tratados con Humira (36.4 %, 29.1 % y 33.7 %, respectivamente) y placebo (7.3 %, 13.0 % y 8.6 %, respectivamente). Se observó un patrón similar de tiempo transcurrido hasta las primeras respuestas ACR 50 y 70 en los cuatro estudios. Algunos pacientes no tratados en forma concomitante con MTX pueden obtener un beneficio adicional si aumentan la frecuencia de administración de Humira a 40 mg cada semana. Esto se confirmó en un estudio a largo plazo de diseño abierto en el que pacientes con una respuesta incompleta aumentaron la frecuencia de administración de 40 mg semana por medio a 40 mg semanalmente. Estudio V en AR: En el estudio V en AR en pacientes con artritis reumatoidea temprana que eran naive para metotrexato, la terapia combinada con Humira y metotrexato condujo a respuestas ACR más rápidas y significativamente mayores que la monoterapia con metotrexato y la monoterapia con Humira en la semana 52 y las respuestas fueron sostenidas hasta la semana 104 (ver tabla 5). En la semana 52 todos los componentes individuales de los criterios de respuesta ACR mejoraron con la terapia Humira/metotrexato y las mejorías se mantuvieron hasta la semana 104. Sobre los dos años de estudio, 48.5 % de los pacientes que recibieron terapia combinada con Humira/metotrexato alcanzaron una mayor respuesta clínica (ACR 70 por seis meses continuos) comparado con 27.2% de pacientes que recibieron monoterapia con metotrexato (p < 0.001) y 24.5% de pacientes que recibieron monoterapia con Humira (p < 0.001).
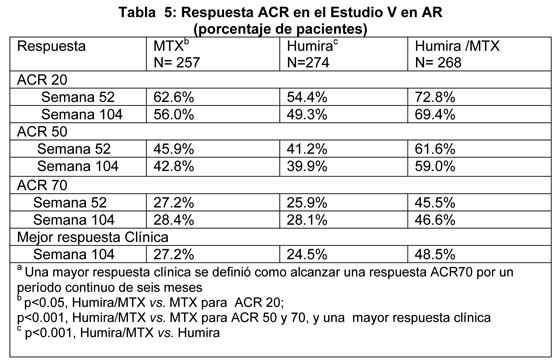
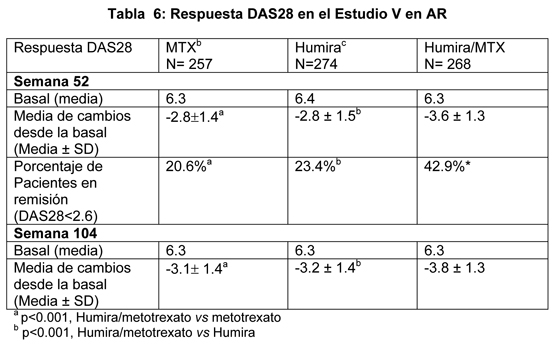
En la semana 52, 42.9% de los pacientes que recibieron terapia combinada Humira/metotrexato alcanzaron remisión clínica (Puntaje de actividad de la enfermedad Disease Activity Score (DAS28) < 2.6) comparado con 20.6% de pacientes que recibieron monoterapia con metotrexato y 23.4% de pacientes que recibieron monoterapia con Humira. La terapia combinada de Humira/metotrexato fue clínica y estadísticamente superior a la monoterapia con metotrexato (p < 0.001) y a la monoterapia con Humira (p < 0.001), en alcanzar un estado bajo de enfermedad en pacientes con artritis reumatoidea moderada a severa recientemente diagnosticada (ver Tabla 6).
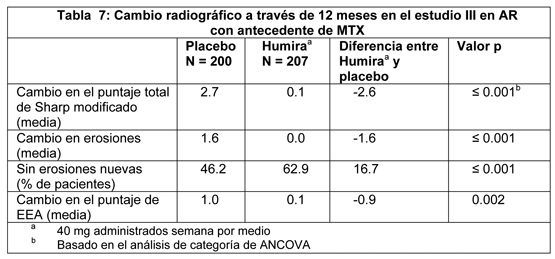
Respuesta radiográfica: En el estudio III en AR, donde los pacientes tratados con Humira tenían una duración media de artritis reumatoidea de aproximadamente 11 años, el daño estructural de la articulación se evaluó radiográficamente y se expresó como cambio en el puntaje de Sharp total modificado y sus componentes, el puntaje de erosión y el puntaje de estrechamiento del espacio articular (EEA). Se evaluaron radiografías de manos/muñecas y del antepié en el período basal, 6 meses y 12 meses. En la tabla 7 se presentan los resultados a los 12 meses. Se observó una diferencia estadísticamente significativa para el cambio en el puntaje de Sharp total modificado (SST) y en el puntaje de erosión a los 6 meses, y se mantuvo a los 12 meses. Los pacientes tratados con Humira/MTX demostraron menos progresión radiográfica que los pacientes que recibieron sólo MTX a las 52 semanas.
En la extensión abierta del estudio III en AR, 77% de los pacientes originales tratados con cualquier dosis de Humira se evaluaron radiográficamente a los 2 años. Los pacientes mantuvieron la inhibición del daño estructural, según lo medido por el SST; cincuenta y cuatro por ciento no tuvo progresión del daño estructural según lo definido por un cambio en el SST de cero o menos. Cincuenta y cinco por ciento (55%) de los pacientes originalmente tratados con Humira 40 mg semana por medio se evaluaron radiográficamente a los 5 años. Los pacientes habían continuado la inhibición del daño estructural con un 50% que no mostraron progresión del daño estructural definido por un cambio en el SST de cero o menos. En el Estudio V en AR, el daño estructural de la articulación se evaluó como en el estudio III en AR. En el grupo con la combinación Humira/metotrexato se observó una mayor inhibición de la progresión radiográfica, evaluada por cambios en el SST, puntaje de erosión y EEA, comparado con el grupo de monoterapia ya sea con metotrexato o con Humira en la semana 52 así como en la semana 104 (ver tabla 8).

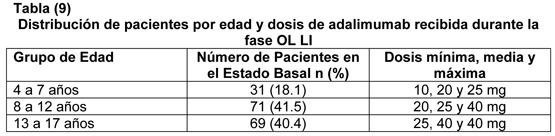
Después de 52 y de 104 semanas de tratamiento, el porcentaje de pacientes sin progresión (cambio de la basal en el puntaje de Sharp total modificado ≤ 0.5) fue significativamente más alto con la terapia combinada Humira/metotrexato (63.8% y 61.2% respectivamente) comparado con la monoterapia de metotrexato (37.4% y 33.5% respectivamente, p < 0.001) y con la monoterapia de Humira (50.7%, p < 0.002 y 44.5%, p < 0.001 respectivamente). Calidad de vida y función física: La calidad de vida relacionada con la salud se evaluó usando el índice de discapacidad del Health Assessment Questionnaire (HAQ) en todos los cinco estudios adecuados y bien controlados, y era un objetivo primario pre-especificado en la semana 52 en el estudio III en AR. Todas las dosis/esquemas de Humira en los cuatro estudios mostraron una mejoría mayor estadísticamente significativa en el índice de discapacidad del HAQ desde el estado basal hasta el mes 6 comparado con placebo. En el estudio III en AR, la mejoría media (CI) en HAQ desde la basal a la semana 52 fue -0,60 (-0,65, -0,55) para los pacientes con Humira/MTX y -0,25 (-0,33, -0,17) para los pacientes con placebo/MTX (p < 0.001). El sesenta y tres por ciento de los pacientes tratados con Humira/MTX alcanzaron una mejoría de 0,5 o más en el HAQ en la semana 52 en la porción doble ciego del estudio. Ochenta y dos por ciento de estos pacientes mantuvo su mejoría hasta la semana 104 y una proporción similar de pacientes mantuvo su respuesta hasta la semana 260 (5 años) del tratamiento abierto. El cuestionario de salud abreviado SF 36 (Short Form Health Survey) también se usó para evaluar la calidad de vida general relacionada con la salud en los cuatro estudios adecuados y bien controlados. Todas las dosis/esquemas de Humira en los cuatro estudios mostraron una mejoría mayor estadísticamente significativa en los puntajes resumidos del componente físico del SF 36 desde el estado basal hasta el mes 6 comparado con placebo, y éste se mantuvo en la semana 52 en el estudio III en AR. La mejoría en el SF 36 también se mantuvo hasta el final de las mediciones en la semana 156 (36 meses). Los puntajes resumidos del componente mental del SF 36 en los estudios II y IV también fueron mayores estadísticamente significativos en el mes 6 para Humira vs. placebo. Los puntajes en los dominios de dolor y vitalidad del SF 36 mostraron una mejoría mayor estadísticamente significativa desde el estado basal hasta el mes 6 en los cuatro estudios para la dosis de 40 mg semana por medio de Humira comparado con placebo. Estos hallazgos se apoyaron por los puntajes de evaluación funcional del tratamiento de enfermedades crónicas FACIT (Functional Assessment of Chronic Illness Therapy), que mostraron una disminución estadísticamente significativa de la fatiga en el mes 6 en los tres estudios analizados, que se mantuvo en la semana 52 en el estudio III en AR. En el estudio V en AR la mejoría en el índice de discapacidad de HAQ y el componente físico del SF 36 fue mayor para el grupo con terapia combinada Humira/metotrexato vs. tanto los grupos con monoterapia con metotrexato como con Humira (p < 0.001) en la semana 52; esta mejoría se mantuvo hasta la semana 104. Artritis Idiopática Juvenil Poliarticular: La seguridad y eficacia de Humira se evaluaron en un estudio multicéntrico, randomizado, doble ciego, grupo paralelo en 171 niños (4 a 17 años de edad) con artritis idiopática juvenil poliarticular (AIJ) en la fase inicial abierta (OL LI), los pacientes se estratificaron en dos grupos, tratados con MTX (metrotrexato) o no tratados con MTX. Los pacientes que estaban en el estrato sin MTX eran ya sea naïve o habían sido retirados de MTX al menos dos semanas previo al estudio de administración de droga. Los pacientes permanecieron con dosis estable de FAMES y/o prednisona (≤ 0.2 mg/kg/día o 10 mg/día máximo). En la fase OL LI, todos los pacientes recibieron 24 mg/m2 hasta un máximo de 40 mg de Humira semana por medio por 16 semanas. La distribución de pacientes por edad y dosis mínima, media y máxima recibida durante la fase OL LI se presenta en la tabla (9).
Los pacientes que demostraron una respuesta ACR 30 Pediátrico en la semana 16 fueron elegibles para ser randomizados en la fase doble ciego (DB) y recibieron ya sea Humira 24 mg/m2 hasta un máximo de 40 mg, o placebo semana por medio por 32 semanas adicionales o hasta que la enfermedad recayera. Los criterios de recaída de enfermedad se definieron como un empeoramiento de ≥ 30% desde el estado basal en ≥ 3 de 6 criterios importantes del ACR Pediátrico, ≥ 2 articulaciones activas y mejoría de > 30% en no más de 1 de los 6 criterios. Después de 32 semanas o en recaída de enfermedad, los pacientes fueron elegibles para reclutarlos en la fase de extensión abierta.
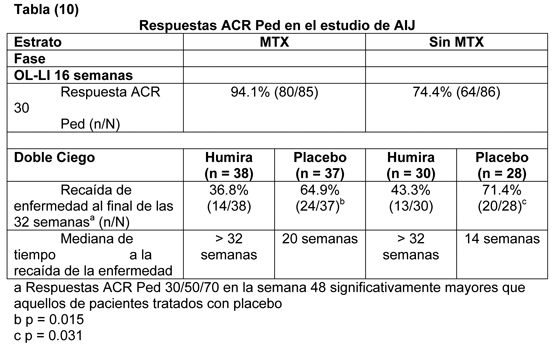
Entre aquellos que respondieron a la semana 16 (n=144), las respuestas ACR Pediátrico 30/50/90 se mantuvieron por hasta dos años en la fase OLE en los pacientes que recibieron Humira a través del estudio. Las respuestas totales fueron generalmente mejores y menos pacientes desarrollaron anticuerpos cuando se trataron con la combinación de Humira y MTX comparado con Humira solo. Tomando estos resultados en consideración, Humira se recomienda para uso en combinación con MTX y para uso como monoterapia en pacientes para quienes el uso de MTX no es apropiado. Estudios clínicos para artritis psoriatica: La seguridad y eficacia de Humira se evaluó en dos estudios aleatorios, doble ciego, placebo-controlados, en 413 pacientes con artritis psoriática. El estudio I en APs (M02-518) enroló 313 pacientes adultos con artritis psoriática moderada a severamente activa ( > 3 articulaciones inflamadas y > 3 articulaciones sensibles) quienes tenían una inadecuada respuesta a la terapia de AINEs en una de las siguientes formas: (1) Compromiso interfalángico distal (DIP) (N=23); (2) artritis poliarticular (ausencia de nódulos reumatoideos y presencia de psoriasis) (N=210); (3) artritis mutilante (N=1); (4) artritis psoriática asimétrica (N=77); o (5) del tipo de espondilitis anquilosante (N=2). Los pacientes en terapia con MTX (158 de 313 pacientes) en el reclutamiento (dosis estable ≤ 30mg/semana por > 1 mes) podían continuar con metotrexato en la misma dosis. Se administraron dosis de Humira 40 mg o placebo semana por medio durante el período doble ciego de 24 semanas del estudio. En el estudio II en APs (M02-570) con 12 semanas de duración, trataron 100 pacientes que tenían una inadecuada respuesta a la terapia con DMARD. Después del término de ambos estudios, 383 pacientes se reclutaron en un estudio de extensión abierto, en que se les administró 40 mg de Humira semana por medio. Respuesta ACR y PASI: Comparado con placebo, el tratamiento con Humira resultó con mejorías en las mediciones de actividad de la enfermedad (ver tablas 11 y 12). Entre los pacientes con artritis psoriática que recibieron Humira, las respuestas clínicas fueron evidentes en algunos pacientes al tiempo de la primera visita (dos semanas). Similares respuestas se observaron en pacientes con cada uno de los subtipos de APs, aunque se reclutaron pocos pacientes con subtipos de artritis mutilante y del tipo de espondilitis anquilosante. Las respuestas fueron similares en los pacientes que estaban o no estaban recibiendo terapia concomitante con MTX en el estado basal. Los pacientes con compromiso psoriático de al menos tres por ciento del área de superficie corporal (ASC) fueron evaluados por las respuestas al Indice de Area Psoriática y Severidad (Psoriatic Area and Severity Index (PASI)). A las 24 semanas, las proporciones de pacientes que alcanzaron una mejoría del 75% o 90% en el PASI eran 59%, y 42% respectivamente, en el grupo con Humira (N=69), comparado con 1%, y 0% respectivamente, en el grupo con placebo (N=69) (p < 0.001). Las respuestas del PASI fueron evidentes en algunos pacientes al momento de la primera visita (dos semanas). Las respuestas fueron similares en los pacientes que estaban o no estaban recibiendo terapia concomitante con MTX en el estado basal.
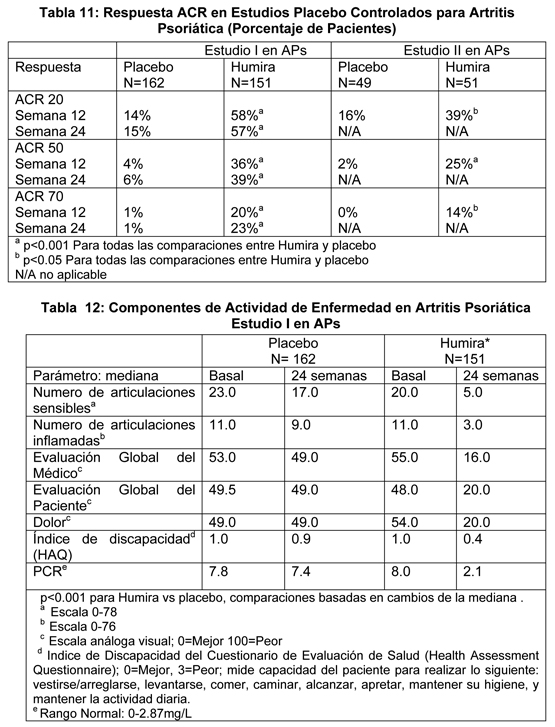
Las respuestas ACR se mantuvieron en el estudio de extensión abierto por hasta 136 semanas. Los cambios radiográficos se evaluaron en los estudios de artritis psoriática. Radiografías de manos, muñecas y pies se obtuvieron en el estado basal y en la semana 24 durante el período doble ciego cuando los pacientes estaban con Humira o placebo y en la semana 48 cuando todos los pacientes estaban en el período abierto con Humira. Se usó un Puntaje Sharp Total (mTSS) modificado, que incluyó articulaciones interfalángicas distales (ej., no idéntico al TSS usado para artritis reumatoidea). El tratamiento con Humira disminuyó la tasa de progresión del daño articular periférico comparado con el tratamiento con placebo medido por cambio desde el estado basal en el puntaje mTSS (media + DS) 0.8 ± 2.5 en el grupo placebo (en la semana 24) comparado con 0.0 ± 1.9 en el grupo Humira (en la semana 48); p < 0.001. En sujetos tratados con Humira sin progresión radiográfica desde el estado basal hasta la semana 48 (n=102), 84% continuaron mostrando no progresión radiográfica a través de las 144 semanas de tratamiento. Calidad de Vida y Función Física: Los pacientes tratados con Humira demostraron mejoría estadísticamente significativa en la función física evaluada por HAQ y Encuesta de Salud en el Formulario Corto (Short Form Health Survey (SF 36)) comparado con placebo en la semana 24. La mejoría de la función física continuó durante la extensión abierta hasta la semana 136. Estudios clínicos para espondilitis anquilosante: La seguridad y eficacia de Humira 40 mg semana por medio se evaluó en 393 pacientes adultos en dos estudios de 24 semanas, placebo-controlados, randomizados, doble ciego, en pacientes con espondilitis anquilosante activa (EA). El estudio más grande (Estudio I en EA o M03-607) enroló a 315 pacientes adultos con EA activa (definido como el cumplimiento de por lo menos dos de los siguientes tres criterios: (1) un puntaje del índice de actividad de enfermedad EA Bath (Bath Ankylosing Spondylitis Disease Activity Index (BASDAI)≥ 4 cm, (2) un puntaje visual análogo (PVA) para el dolor de espalda total ≥40 mm, (3) rigidez matutina ≥1 hora), quienes tenían una respuesta inadecuada a la terapia convencional. El período ciego es seguido por un período abierto durante el cual los pacientes recibieron Humira 40 mg subcutáneo semana por medio por hasta 28 semanas adicionales. Los resultados mostraron una mejoría estadísticamente significativa de los signos y síntomas de EA en los pacientes tratados con Humira comparado con placebo. La mejoría significativa en las mediciones de actividad de enfermedad, se observó primero en la semana 2 y se mantuvo hasta las 24 semanas como se muestra en la figura 4 y las tablas 13 y 14. Se incluyeron pacientes con anquilosis espinal total en este estudio (n=11). Las respuestas de estos pacientes fueron similares a aquellas sin anquilosis total.
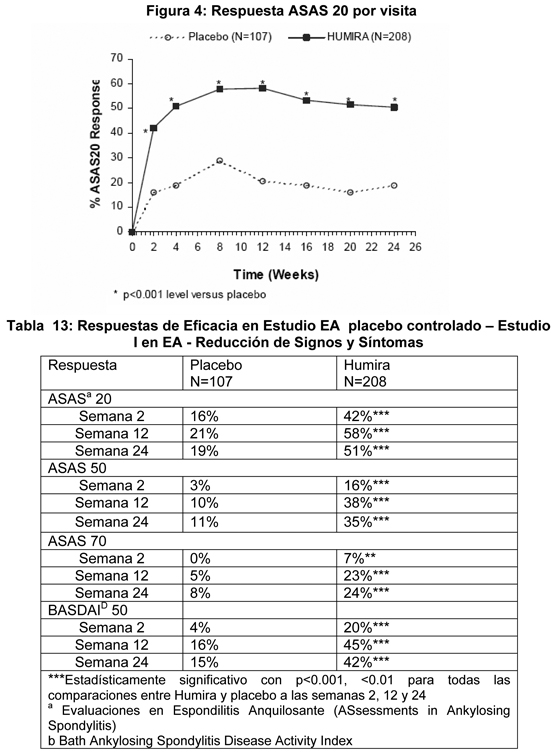
Se alcanzó un bajo nivel de actividad de enfermedad (definido como un valor < 20 (en la escala de 0 a 100 mm) en cada uno de los cuatro parámetros de respuesta ASAS) a las 24 semanas en un 22% de los pacientes tratados con Humira vs. 6% de los pacientes tratados con placebo (p < 0.001).
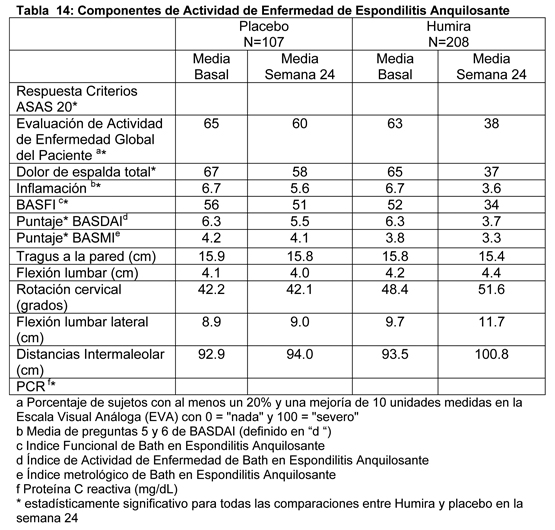
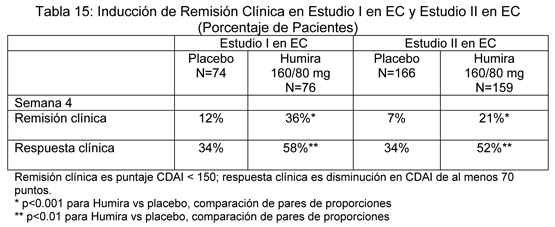
Tendencias similares se observaron en un estudio más pequeño, randomizado, doble ciego, placebo controlado (Estudio II en EA, M03-606) de 82 pacientes adultos con espondilitis anquilosante activa. Los resultados informados de los pacientes se evaluaron en ambos estudios para espondilitis anquilosante usando el cuestionario genérico de estado de salud SF-36 y el específico de enfermedad Cuestionario de Calidad de Vida en Espondilitis Anquilosante (Ankylosing Spondylitis Quality of Life Questionnaire (ASQoL)). Los pacientes tratados con Humira tenían una mejoría significativamente mayor en el Puntaje del Componente Físico del SF-36 (cambio medio 6.93) comparado a los pacientes tratados con placebo (cambio medio 1.55; p < 0.001) en la semana 12, el cual se mantuvo hasta la semana 24 (cambio medio 7.44 vs. 1.85). Los resultados del ASQoL apoyan estos hallazgos que demuestran mejoría en la calidad de vida global. Los pacientes tratados con Humira tuvieron una mejoría estadísticamente significativa (cambio medio - 3.15) comparada con los pacientes tratados con placebo (cambio medio - 0.95; p < 0.001) en la semana 12, la cual se mantuvo hasta la semana 24 (cambio medio -3.58 vs. -1.06). Estudios clínicos para enfermedad de Crohn: La seguridad y eficacia de múltiples dosis de Humira se evaluaron en pacientes adultos con enfermedad de Crohn (EC) moderada a severamente activa (Indice de Actividad de Enfermedad de Crohn (CDAI) ≥220 y ≤450) en estudios randomizados, doble ciego, placebo controlados. Se permitieron dosis estables concomitantes de aminosalicilatos, corticosteroides y/o agentes inmunomoduladores, y 79% de los pacientes continuaron recibiendo al menos uno de estos medicamentos. La inducción de la remisión clínica (definida como CDAI < 150) se evaluó en dos estudios. En el Estudio I en EC (CLASSIC I), 299 pacientes naive a bloqueadores-TNF se randomizaron para uno de cuatro grupos de tratamiento: el grupo placebo recibió placebo en las Semanas 0 y 2, el grupo 160/80 recibió 160 mg de Humira en la Semana 0 y 80 mg en la Semana 2, el grupo 80/40 recibió 80 mg en la Semana 0 y 40 mg en la Semana 2, y el grupo 40/20 recibió 40 mg en la Semana 0 y 20 mg en la Semana 2. Los resultados clínicos se evaluaron en la Semana 4. En el segundo estudio de inducción, Estudio II en EC (GAIN), 325 pacientes que habían perdido respuesta a, o eran intolerantes a, terapia previa con infliximab se randomizaron para recibir ya sea 160 mg de Humira en la Semana 0 y 80 mg en la Semana 2, o placebo en las Semanas 0 y 2. Los resultados clínicos se evaluaron en la Semana 4. La mantención de la remisión clínica se evaluó en el Estudio III en EC (CHARM). En este estudio, 854 pacientes con enfermedad activa recibieron Humira como estudio abierto, 80 mg en la Semana 0 y 40 mg en la Semana 2. Los pacientes luego se randomizaron en la Semana 4 a 40 mg de Humira semana por medio, 40 mg de Humira cada semana, o placebo. La duración total del estudio fue 56 semanas. Los pacientes con respuesta clínica (disminución en CDAI ≥70) en la Semana 4 se estratificaron y analizaron separadamente de aquellos que no tenían respuesta clínica en la Semana 4. Inducción de Remisión Clínica: Un mayor porcentaje de pacientes tratados con 160/80 mg de Humira alcanzaron inducción de remisión clínica versus placebo en la Semana 4 sin importar si los pacientes eran naive a bloqueadores-TNF (Estudio I en EC), o habían perdido respuesta a o eran intolerantes a infliximab (Estudio II en EC) (ver Tabla 15).
Mantención de Remisión Clínica: En el estudio III en EC en la Semana 4, 58% (499/854) de los pacientes estaban con respuesta clínica y se evaluaron en el análisis primario. En las Semanas 26 y 56, mayor proporción de pacientes que estaban con respuesta clínica en la Semana 4 alcanzaron remisión clínica en el grupo con Humira 40 mg semana por medio comparado con los pacientes en el grupo con placebo (ver Tabla 16). El grupo que recibió terapia con Humira cada semana no demostró mayores tasas de remisión significativas comparado con el grupo que recibió Humira semana por medio.
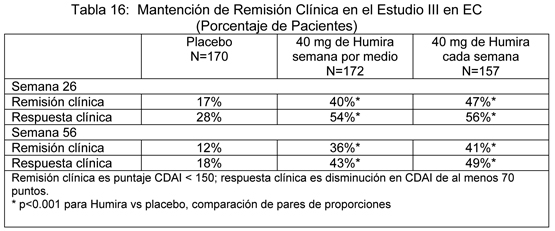
De aquellos con respuesta en la Semana 4 que lograron remisión durante el estudio, los pacientes en el grupo de Humira semana por medio mantuvieron la remisión por un tiempo mayor que los pacientes en el grupo de con placebo. El uso de Humira en enfermedad de Crohn de más de un año no se ha evaluado en estudios clínicos controlados. Calidad de Vida: En el Estudio I y Estudio II en EC, se alcanzó una mejoría estadísticamente significativa en el puntaje total del cuestionario de enfermedad específico para enfermedad inflamatoria del intestino (IBDQ), en la semana 4 en los pacientes randomizados con Humira 80/40 mg y 160/80 mg comparado con placebo y en las semanas 26 y 56 en el Estudio III en EC se observó también entre los grupos de tratamiento con adalimumab comparado al grupo placebo. Estudios clínicos para psoriasis: La seguridad y eficacia de Humira se estudiaron en pacientes adultos con psoriasis crónica en placa (≥10% BSA comprometido e Indice de Severidad y Area de Psoriasis (PASI - Psoriasis Area and Severity Index) ≥12 o ≥10), que eran candidatos a terapia sistémica o fototerapia en estudios randomizados, doble ciego. 73% de los pacientes reclutados en los Estudios I y II de Psoriasis habían recibido terapia sistémica o fototerapia previa. El Estudio I de Psoriasis (REVEAL) evaluó 1212 pacientes dentro de tres períodos de tratamiento. En el período A, los pacientes recibieron placebo o Humira en una dosis inicial de 80 mg seguido por 40 mg semana por medio comenzando una semana después de la dosis inicial. Después de 16 semanas de terapia, los pacientes que alcanzaron al menos una respuesta PASI 75 (puntaje PASI de mejoría de al menos 75% relativo al estado basal), entraron al período B y recibieron Humira 40 mg en forma abierta semana por medio.
Los pacientes que mantuvieron respuesta ≥PASI 75 en la semana 33 y estaban originalmente randomizados en terapia activa en el Período A, se re-randomizaron en el período C para recibir 40 mg de Humira semana por medio o placebo por un adicional de 19 semanas. A través de todos los grupos de tratamiento, la media del estado basal del puntaje PASI fue 18.9 y la Evaluación Global del Médico (PGA - Physician's Global Assessment) en el estado basal varió desde "moderado" (53% de los sujetos incluidos) a "severo" (41%) a "muy severo" (6%). El Estudio II de Psoriasis (CHAMPION) comparó la eficacia y seguridad de Humira versus metotrexato y placebo en 271 pacientes. Los pacientes recibieron placebo, una dosis inicial de MTX de 7.5 mg y después aumentos de dosis hasta la semana 12, con una dosis máxima de 25 mg o una dosis inicial de 80 mg de Humira seguido por 40 mg semana por medio (comenzando una semana después de la dosis inicial) por 16 semanas. No existen datos disponibles que comparen Humira y MTX más allá de 16 semanas de terapia. Los pacientes que recibían MTX que alcanzaron una respuesta ≥PASI 50 en la semana 8 y/o 12 no recibieron más aumentos de dosis. A través de todos los grupos de tratamiento, la media del puntaje PASI del estado basal fue 19.7 y el puntaje PGA del estado basal varió desde "leve" ( < 1%) a "moderado" (48%) a "severo" (46%) a "muy severo" (6%). En los estudios I y II de Psoriasis, un objetivo primario era la proporción de pacientes que alcanzaron una respuesta PASI 75 desde el estado basal a la semana 16 (ver tablas 17 y 18).
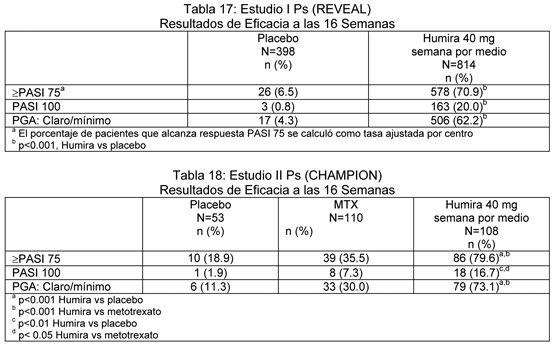
En el Estudio I de Psoriasis, 28% de los pacientes que eran respondedores PASI 75 y fueron re-randomizados a placebo en la semana 33 comparado al 5% que continuaron con Humira, p < 0.001, presentaron "pérdida de respuesta adecuada" (puntaje PASI después de la semana 33 y después o antes de la semana 52 con una respuesta < PASI 50 relativa al estado basal con un mínimo de 6-puntos de incremento en el puntaje PASI relativo a la semana 33). De los pacientes que perdieron respuesta adecuada después de la re-randomización a placebo que luego se reclutaron dentro del estudio de extensión abierto, 38% (25/66) y 55% (36/66) re-ganaron respuesta PASI 75 después de 12 y 24 semanas de tratamiento, respectivamente. Se demostraron mejorías significativas en la semana 16 desde el estado basal comparado con placebo (Estudios I y II) y MTX (Estudio II) en el DLQI (Dermatology Life Quality Index - Indice Dermatológico de Calidad de Vida). En el Estudio I, los puntajes que resumen las mejorías en el componente físico y mental del SF-36 también fueron significativos comparados con placebo. En un estudio de extensión abierto, para pacientes que escalaron dosis desde 40 mg semana por medio a 40 mg semanal debido a una respuesta PASI bajo 50 y que se evaluaron a las 12 semanas después del escalamiento de dosis, 59/243 (24.3%) de los pacientes alcanzaron respuesta PASI 75. Inmunogenicidad: La formación de anticuerpos anti-adalimumab se asocia con aumento del clearance y reducción de la eficacia de adalimumab. No existe correlación aparente entre la presencia de anticuerpos anti-adalimumab y eventos adversos. Los pacientes en los Estudios I, II y III en AR se analizaron en múltiples puntos de tiempo para anticuerpos para adalimumab durante el período de 6 a 12 meses. En los estudios pivotales, se identificaron anticuerpos anti-adalimumab en 58/1053 (5.5%) de los pacientes tratados con adalimumab, comparado con 2/370 (0.5%) con placebo. En los pacientes que no recibían metotrexato concomitante, la incidencia fue 12.4%, comparado a 0.6% cuando adalimumab se usó como agregado a metotrexato. En pacientes con artritis psoriática, se identificaron anticuerpos de adalimumab en 38/376 sujetos (10%) tratados con adalimumab. En los pacientes que no recibían metotrexato concomitante, la incidencia fue 13.5% (24/178 sujetos), comparado a 7% (14 de 198 sujetos) cuando adalimumab se usó como agregado a metotrexato. En pacientes con espondilitis anquilosante se identificaron anticuerpos en 17/204 sujetos (8.3%) tratados con adalimumab. En los pacientes que no recibían metotrexato concomitante, la incidencia fue 16/185 (8.6%), comparado a 1/19 (5.3%) cuando adalimumab se usó como agregado a metotrexato. En pacientes con enfermedad de Crohn, se identificaron anticuerpos de adalimumab en 7/269 sujetos (2.6%) tratados con adalimumab. En pacientes con psoriasis, se identificaron anticuerpos anti-adalimumab en 77/920 sujetos (8.4%) tratados con monoterapia con adalimumab. En pacientes con artritis idiopática juvenil, se identificaron anticuerpos anti-adalimumab en 15.8% de los pacientes tratados con Humira. En pacientes que recibieron metotrexato concomitante, la incidencia fue 5.9% comparado a 25.6% con monoterapia con Humira. Debido a que los análisis de inmunogenicidad son producto-específicos, no es apropiado hacer comparaciones de tasas de anticuerpos con aquellas de otros productos.
Indicaciones.
Artritis Reumatoidea: Humira se indica para reducir los signos y síntomas, incluyendo mejoría en la respuesta clínica y remisión clínica inhibiendo la progresión del daño estructural y mejorando la función física en pacientes adultos con artritis reumatoidea moderada a severamente activa (AR). Humira ha mostrado reducir la tasa de progresión del daño articular medido por rayos X y mejora la función física cuando se administra junto con metotrexato. Humira se puede utilizar solo o en combinación con metotrexato u otras drogas antirreumáticas modificadoras de enfermedad (DMARDs). Artritis Psoriática: Humira se indica para reducir los signos y síntomas de artritis activa en pacientes con artritis psoriática (APs). Humira ha mostrado que reduce la tasa de progresión de daño articular periférico estructural y mejora la función física. Humira se puede utilizar solo o en combinación con DMARDs. Espondilitis Anquilosante: Humira se indica para reducir los signos y síntomas en pacientes con Espondilitis Anquilosante (EA) activa que tienen una respuesta inadecuada a la terapia convencional. Enfermedad de Crohn: Humira se indica para reducir los signos y síntomas, e inducir y mantener la remisión clínica en pacientes adultos con enfermedad de Crohn (EC) moderada a severamente activa que han tenido una respuesta inadecuada a la terapia convencional. Humira se indica para reducir los signos y síntomas e inducir remisión clínica en estos pacientes si ellos también han perdido respuesta a o son intolerantes a infliximab. Psoriasis en Placa: Humira se indica para el tratamiento de pacientes adultos con psoriasis crónica en placa moderada a severa que son candidatos a terapia sistémica o fototerapia y cuando otras terapias sistémicas son médicamente menos apropiadas. Artritis Idiopática Juvenil Poliarticular: Humira se indica para reducir los signos y síntomas de la artritis idiopática juvenil poliarticular (AIJ) moderada a severamente activa en pacientes de 13 a 17 años de edad. Humira se puede usar solo o en combinación con metotrexato.
Dosificación.
Artritis Reumatoidea, Artritis Psoriática y Espondilitis Anquilosante: La dosis recomendada de Humira para pacientes adultos con artritis reumatoidea, artritis psoriática o espondilitis anquilosante es 40 mg administrados semana por medio, como dosis única vía inyección subcutánea. Durante el tratamiento con Humira, se puede continuar la administración de metotrexato, glucocorticoides, salicilatos, antiinflamatorios no esteroidales, analgésicos u otros DMARDs. En artritis reumatoidea, algunos pacientes no tratados con MTX de forma concomitante podrían obtener un beneficio adicional si aumentan la frecuencia de administración de Humira a 40 mg cada semana (opcional). Enfermedad de Crohn: El régimen de dosificación inicial, recomendado para pacientes con enfermedad de Crohn severa, es de 80 mg el Día 1 seguido por 40 mg dos semanas después (cada 15). En caso de requerirse una respuesta más rápida al tratamiento se pueden administrar 160 mg inicialmente en el Día 1 (administrado como cuatro inyecciones de 40 mg en un día o como dos inyecciones de 40 mg por día por dos días consecutivos), seguido por 80 mg dos semanas después (Día 15), debiendo tenerse en cuenta el mayor riesgo de reacciones adversas durante el inicio del tratamiento. Otras dos semanas después (Día 29) iniciar una dosis de 40 mg semana por medio. Durante el tratamiento con Humira, se puede continuar la administración de aminosalicilatos, corticosteroides y/o agentes inmunomoduladores (ej. 6-mercaptopurina y azatioprina). Algunos pacientes que experimentan disminución en su respuesta se pueden beneficiar de un aumento en la intensidad de la dosis a 40 mg de Humira cada semana. Algunos pacientes que no han respondido en la semana 4 se pueden beneficiar de una terapia continua hasta la semana 12. La terapia continua se debe reconsiderar cuidadosamente en un paciente que no responde dentro de este período de tiempo. Durante el tratamiento de mantención, los corticosteroides se pueden disminuir de acuerdo con las guías de práctica clínica. Psoriasis en Placa: La dosis recomendada de Humira para pacientes adultos con psoriasis en placa es una dosis inicial de 80 mg, seguida por 40 mg administrados semana por medio comenzando una semana después de la dosis inicial. En pacientes que no responden, una terapia continua de más de 16 semanas debería ser cuidadosamente evaluada. La administración más allá de 1 año en esta indicación no ha sido evaluada en estudios clínicos controlados. Artritis Idiopática Juvenil Poliarticular: La dosis recomendada de Humira pacientes con artritis idiopática juvenil poliarticular de 13 a 17 años de edad es 40 mg de adalimumab administrados semana por medio como una dosis única vía inyección subcutánea. Los datos sugieren que la respuesta clínica se alcanza generalmente dentro de las 12 semanas de tratamiento. La terapia continuada se debe reconsiderar cuidadosamente en un paciente que no responde en este período de tiempo. Preparación de Humira: Humira se debe usar bajo la dirección y supervisión de un médico. Los pacientes se pueden autoinyectar Humira si su médico considera que es apropiado, con un seguimiento médico según sea necesario después de un apropiado entrenamiento en la técnica de inyección subcutánea. Los sitios de auto-inyección incluyen el muslo o el abdomen. Los sitios de inyección deben ser rotados. Nunca deben administrarse nuevas inyecciones en zonas en las que la piel presente dolor, hematoma, enrojecimiento o endurecimiento. Los productos farmacológicos parenterales se deben inspeccionar visualmente en busca de partículas sólidas o cambio de coloración previo a su administración, siempre que la solución y el envase lo permitan. Humira no se debe mezclar en la misma jeringa o vial con ningún otro medicamento. Todo el producto no utilizado o el material de desecho se deben eliminar conforme a los requisitos locales. Uso Pediátrico: Humira no se ha estudiado en niños menores de 4 años de edad y existen datos limitados del tratamiento con Humira en niños con peso < 15 kg. La seguridad y eficacia de Humira en pacientes pediátricos para otras indicaciones aparte de artritis idiopática juvenil no se han establecido. Uso Geriátrico: Del número total de sujetos en los estudios clínicos de Humira, el 12% tenía 65 años de edad o más, mientras que aproximadamente el 2.5% tenía 75 años de edad o más. No se observaron diferencias globales con respecto a la efectividad entre estos sujetos y los sujetos más jóvenes. No obstante, debido a que existe una mayor incidencia de infecciones en los adultos mayores respecto a la población general, se debe tener precaución cuando se trata a adultos mayores. No es necesario hacer ajuste de dosis para esta población.
Contraindicaciones.
Humira no se debe administrar a pacientes con hipersensibilidad conocida a Humira o a cualquiera de sus excipientes.
Embarazo y lactancia.
Se ha realizado un estudio de desarrollo de toxicidad embrio-fetal perinatal en monos cynomolgus con dosis de hasta 100 mg/kg (266 veces el ABC en el ser humano en dosis de 40 mg sc con MTX cada semana, o 373 veces en dosis de 40 mg sc sin MTX) que no ha revelado evidencia de daño a los fetos debido a adalimumab. No obstante, no se han realizado estudios adecuados y bien controlados en mujeres embarazadas. Debido a que los estudios en reproducción y desarrollo animal no son siempre predictivos de respuesta humana, HUMIRA se debe utilizar en el embarazo sólo en caso claramente necesario. No existen datos clínicos disponibles de adalimumab en mujeres embarazadas. Por consiguiente, no se debe administrar HUMIRA durante el embarazo a menos que sea claramente necesario. A las mujeres en edad fértil se debe recomendar que no queden embarazadas durante el tratamiento con HUMIRA. Trabajo de Parto y Parto: No existen efectos conocidos de Humira sobre el trabajo de parto o el parto. Madres en Lactancia: No se sabe si adalimumab se excreta en la leche materna ni si se absorbe por vía sistémica tras su ingestión. Debido a que muchos fármacos e inmunoglobulinas se excretan en la leche materna y el potencial de reacciones adversas serias de Humira en lactantes, se debe decidir si interrumpir la lactancia natural o suspender el fármaco, teniendo en cuenta la importancia de éste para la madre.
Reacciones adversas.
Estudios Clínicos de Artritis Reumatoidea, Artritis Idiopática Juvenil Poliarticular, Artritis Psoriática, Espondilitis Anquilosante, Enfermedad de Crohn y Psoriasis: Humira se estudió en 6593 pacientes en estudios controlados y abiertos por hasta 60 meses. Estos estudios incluyeron pacientes con artritis reumatoidea con enfermedad de corta y de larga data, artritis idiopática juvenil poliarticular, así como pacientes con artritis psoriática, espondilitis anquilosante, enfermedad de Crohn y psoriasis. Los datos presentados a continuación se basan en los estudios controlados pivotales que involucran 4355 pacientes que reciben Humira y 2487 pacientes que reciben placebo o comparador activo durante el período controlado. La proporción de pacientes que discontinuaron el tratamiento debido a eventos adversos durante la fase doble ciego, controlada de los estudios pivotales fue 4.5% para los pacientes tratados con Humira y 4.5% para los pacientes control. Se puede esperar que aproximadamente 15% de los pacientes presenten reacciones en el sitio de inyección basado en uno de los eventos adversos más comunes con adalimumab en estudios clínicos controlados. En la tabla 1 mostrada a continuación se presentan los eventos adversos tanto clínicos como de laboratorio, con una relación de causalidad con adalimumab al menos posible, por sistema órgano clase y por frecuencia (muy común ≥1/10; común ≥1/100 a ≤1/10; poco común ≥1/1000 a < 1/100; raro ≥1/10000 a < 1/1000). Se ha incluido la frecuencia más alta observada entre las variadas indicaciones. Un asterisco (*) aparece en la columna CSO si mayor información se encuentra en otro lugar en las secciones Contraindicaciones, Advertencias y Precauciones y Reacciones Adversas.
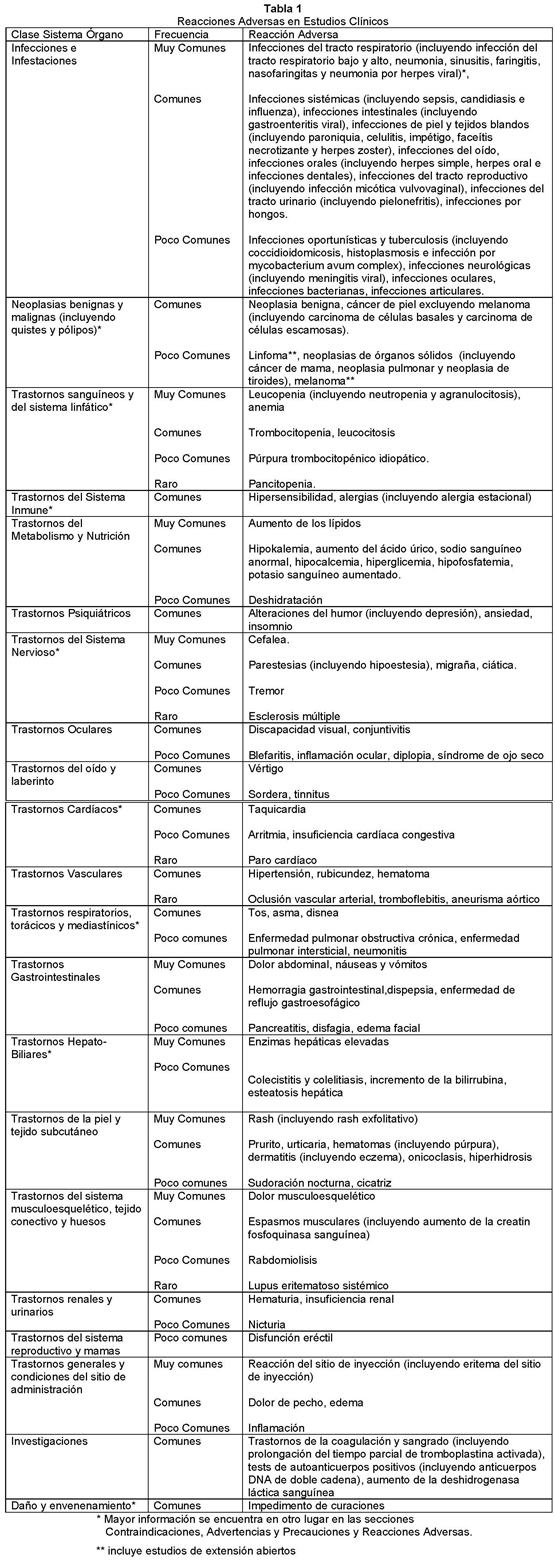
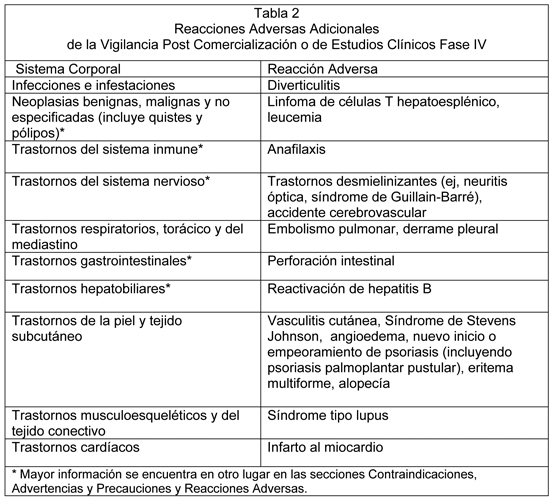
Población Pediátrica: En general, las reacciones adversas en pacientes pediátricos fueron similares en frecuencia y tipo a aquellos observados en pacientes adultos. Reacciones en el sitio de inyección: En los estudios controlados pivotales, 15 % de los pacientes tratados con Humira desarrolló reacciones en el sitio de inyección (eritema y/o prurito, hemorragia, dolor o tumefacción) en comparación con 9 % de los pacientes que recibieron tratamiento control. La mayoría de las reacciones en el sitio de inyección se describieron como leves y generalmente no necesitaron discontinuar el fármaco. Infecciones: En los estudios controlados pivotales, la tasa de infección fue de 1.58 por paciente año en los pacientes tratados con Humira y de 1.42 por paciente año en los pacientes control. La incidencia de infecciones graves fue de 0.03 por paciente año en los pacientes tratados con Humira y de 0.04 por paciente año en los pacientes control. Las infecciones fueron principalmente infecciones de las vías respiratorias superiores, bronquitis e infecciones de las vías urinarias. La mayoría de los pacientes continuó el tratamiento con Humira tras la resolución de la infección. En estudios controlados y abiertos con Humira, se han informado infecciones serias (incluyendo infecciones fatales, que ocurrieron raramente), las cuales incluyen reportes de tuberculosis (incluyendo TBC miliar y ubicaciones extrapulmonares) e infecciones invasivas oportunistas (ej. histoplasmosis diseminada, neumonía por pneumocistis carinii, aspergilosis y listeriosis). Neoplasias malignas y Trastornos Linfoproliferativos: No se observaron neoplasias malignas en 171 pacientes con una exposición de 192.5 pacientes año durante un estudio de Humira en pacientes con artritis idiopática juvenil. Durante las fases controladas de los estudios pivotales de Humira de al menos 12 semanas de duración en pacientes con artritis reumatoidea moderada a severamente activa, artritis psoriática, espondilitis anquilosante, enfermedad de Crohn y psoriasis, neoplasias malignas, otros distintos de linfoma y cáncer de piel de tipo no melanoma, se observaron en una tasa (intervalo de confianza 95%) 5.9 (3.5, 9.9) por 1000 pacientes-año entre 3853 pacientes tratados con Humira vs. una tasa de 4.3 (1.8, 10.4) por 1000 pacientes-año entre 2183 pacientes control (mediana de duración del tratamiento fue de 5.5 meses para Humira y 3.9 meses para los pacientes tratados con control). La tasa (intervalo de confianza 95%) de cánceres de piel tipo no melanoma fue 8.8 (5.7, 13.5) por 1000 pacientes-año entre los pacientes tratados con Humira y 2.6 (0.8, 8.0) por 1000 pacientes-año entre pacientes control. De estos cánceres de piel, los carcinomas de células escamosas ocurrieron en tasas (intervalo de confianza 95%) de 2.5 (1.1, 5.6) por 1000 pacientes-año entre los pacientes tratados con Humira y 0.9 (0.1, 6.1) por 1000 pacientes-año entre los pacientes control. La tasa (intervalo de confianza 95%) de linfomas fue 0.8 (0.2, 3.3) por 1000 pacientes-año entre pacientes tratados con Humira y 0.9 (0.1, 6.1) por 1000 pacientes-año entre pacientes control. La tasa observada de neoplasias malignas, diferentes a linfoma y cánceres de piel no melanoma, es aproximadamente 8.3 por 1000 pacientes año en la porción controlada de los estudios clínicos y en los estudios de extensión abiertos en curso. La tasa observada de cánceres de piel no melanoma es aproximadamente 9.3 por 1000 pacientes año y la tasa observada de linfomas es aproximadamente 1.2 por 1000 pacientes año. La mediana de duración de estos estudios es aproximadamente 2.7 años e incluyeron 4.767 pacientes que estuvieron con Humira por al menos 1 año o que desarrollaron una neoplasia maligna dentro del año de inicio de terapia, representando sobre 15.332 pacientes año de terapia. Autoanticuerpos: En los estudios I-V en AR, se realizaron análisis de autoanticuerpos en las muestras de suero de pacientes en múltiples puntos de tiempo. En estos estudios adecuados y bien controlados, 11.9% de los pacientes tratados con Humira y 8.1% de los pacientes tratados con placebo y control activo, que tenían títulos de anticuerpos antinucleares negativos en el estado basal reportaron títulos positivos en la semana 24. Dos pacientes fuera de los 3989 tratados con Humira en todos los estudios para AR, APs y EA, desarrollaron signos clínicos sugerentes de un nuevo inicio de un síndrome tipo lupus. Los pacientes mejoraron después de la discontinuación de la terapia. Ningún paciente desarrolló nefritis lúpica ni presentó síntomas de sistema nervioso central. El impacto del tratamiento a largo plazo con Humira sobre el desarrollo de enfermedades autoinmunes se desconoce. Psoriasis: Nuevo inicio o Empeoramiento: Casos de nuevos inicios de psoriasis, incluyendo psoriasis pustular y psoriasis palmoplantar, y casos de empeoramiento de psoriasis pre-existente se han reportado con el uso de bloqueadores de TNF, incluyendo Humira. Muchos de estos pacientes estaban tomando inmunosupresores concomitantes (ej, MTX, corticosteroides). Algunos de estos pacientes habían mejorado su psoriasis después de la discontinuación de su bloqueador de TNF. Algunos pacientes habían tenido recurrencias de la psoriasis cuando ellos fueron re-tratados con un bloqueador de TNF diferente. Se debe considerar la discontinuación de Humira para los casos severos y aquellos que no mejoran o empeoran a pesar de los tratamientos tópicos. Elevación de las Enzimas Hepáticas: Estudios clínicos en artritis reumatoidea: en estudios clínicos controlados en artritis reumatoidea (Estudios en AR I-IV), las elevaciones de ALT fueron similares en los pacientes que recibían adalimumab o placebo. En pacientes con artritis reumatoidea temprana (duración de enfermedad de menos de 3 años) (Estudio V), las elevaciones de ALT fueron más comunes en el brazo combinado (Humira/metotrexato) comparado con el brazo de monoterapia con metotrexato o el brazo de monoterapia con Humira. Estudios clínicos en artritis idiopática juvenil poliarticular: En el estudio de AIJ, las pocas elevaciones de transaminasas fueron pequeñas y similares en los pacientes expuestos a placebo y adalimumab y mayormente ocurrieron en combinación con metotrexato. Estudios clínicos en artritis psoriática: las elevaciones de ALT fueron más comunes en pacientes con artritis psoriática (Estudios en APs I-II) comparado con pacientes en estudios clínicos de artritis reumatoidea. En todas las indicaciones los pacientes con ALT elevadas fueron asintomáticos y en la mayoría de los casos las elevaciones eran transitorias y se resolvieron con la continuación del tratamiento. Eventos Adversos Adicionales de la Vigilancia Post Comercialización o Estudios Clínicos Fase IV: Se han reportado eventos adversos durante el uso después de la aprobación de Humira. Debido a que estos eventos son reportados voluntariamente desde una población de tamaño indeterminado, no es siempre posible estimar confiablemente su frecuencia o establecer una relación causal a la exposición de Humira.
Advertencias.
Infecciones: Infecciones serias, debido a bacterias, micobacterias, hongos invasivo (histoplasmosis diseminada o extrapulmonar, aspergillosis, coccidioidomicosis), virales, parasitarias u otras infecciones oportunísticas se han reportado en pacientes que reciben agentes antagonistas de TNF. Sepsis, casos raros de tuberculosis, candidiasis, listeriosis y pneumocystis también se han informado con el uso de antagonistas TNF, incluyendo Humira. Otras infecciones serias observadas en los estudios clínicos incluyen pneumonia, pielonefritis, artritis séptica y septicemia. Se han informado hospitalizaciones o resultados fatales asociados con infecciones. Muchas de estas infecciones serias han ocurrido en pacientes que estaban recibiendo en forma concomitante terapia inmunosupresora, que además de su enfermedad subyacente, podría predisponerlos a infecciones. El tratamiento con Humira no se debe iniciar en pacientes con infecciones activas, incluidas las infecciones crónicas o localizadas, hasta que éstas estén controladas. En pacientes que han sido expuestos a la tuberculosis, y pacientes que han viajado en áreas de alto riesgo de tuberculosis o micosis endémica, tales como histoplasmosis, coccidioidomicosis, o blastomicosis, el riesgo y beneficios del tratamiento con Humira se debe considerar previo al inicio de la terapia (ver Otras Infecciones Oportunísticas). Como en el caso de otros antagonistas TNF, se debe vigilar estrechamente a los pacientes con respecto al desarrollo de infecciones, incluida la tuberculosis, antes, durante y hasta 5 meses después del tratamiento con Humira. Los pacientes que desarrollan una nueva infección durante el tratamiento con Humira se deben controlar estrechamente y someter a una completa evaluación diagnóstica. La administración de Humira se debe discontinuar si el paciente desarrolla una nueva infección seria o sepsis, y se debe iniciar una apropiada terapia antimicrobiana o antifúngica hasta que las infecciones estén controladas. Los médicos deben tener precaución al considerar el uso de Humira en pacientes con historia de infecciones recurrentes o con condiciones subyacentes que puedan predisponer a los pacientes al desarrollo de infecciones. Tuberculosis: Al igual que con otros antagonistas-TNF, se ha informado tuberculosis (presentación clínica frecuentemente diseminada o extrapulmonar) asociada con la administración de Humira en estudios clínicos. Aunque se observaron casos con todas las dosis, la incidencia de reactivación de tuberculosis se aumentó particularmente con dosis de Humira que eran superiores a la dosis recomendada. Antes de iniciar el tratamiento con Humira, en todos los pacientes se debe evaluar tanto la infección por tuberculosis activa como inactiva (latente). Esta evaluación debe incluir una detallada historia médica con la posible exposición previa a pacientes con tuberculosis activa y tratamiento inmunosupresor previo y/o actual. Se deben realizar pruebas de screening apropiadas (p. ej., radiografía de tórax o prueba de la tuberculina) de acuerdo a las recomendaciones locales. El tratamiento de infecciones latentes de tuberculosis se debe iniciar previo a la terapia con Humira. Cuando el test de tuberculina de piel se realiza para infección latente de tuberculosis, una induración de 5 mm de tamaño o mayor se debe considerar positiva, aunque haya sido previamente vacunado con el Bacilo de Calmette-Guerin (BCG). Se debe considerar la posibilidad de una tuberculosis latente no detectada especialmente en pacientes que han inmigrado de o viajaron a países con una alta prevalencia de tuberculosis o que han tenido contacto cercano con una persona con tuberculosis activa. Si se diagnostica tuberculosis activa, no se debe iniciar terapia con Humira. Si se diagnostica una tuberculosis latente, se debe iniciar una apropiada profilaxis anti-tuberculosis conforme con las recomendaciones locales antes de iniciar el tratamiento con Humira. Se debe considerar la terapia anti-tuberculosis previo al inicio de Humira en pacientes que tienen un test negativo para tuberculosis latente pero tienen factores de riesgo para infección por TBC. La decisión de iniciar la terapia anti-tuberculosis en estos pacientes sólo se debe hacer después de tomar en consideración tanto el riesgo para infección latente por tuberculosis como los riesgos de la terapia anti-tuberculosis. Si es necesario, se debe consultar a un médico con experiencia en el tratamiento de tuberculosis. El tratamiento anti-tuberculosis de pacientes con infección latente por tuberculosis disminuye el riesgo de reactivación en pacientes que reciben tratamiento con Humira. Sin embargo, se ha desarrollado tuberculosis activa en pacientes que reciben Humira en quienes el screening para infección latente por tuberculosis fue negativo, y algunos pacientes que previamente habían recibido tratamiento para tuberculosis latente o activa han desarrollado tuberculosis activa mientras estaban siendo tratados con agentes bloqueadores de TNF. Los pacientes que reciben Humira se deben monitorear por signos y síntomas de tuberculosis activa, particularmente porque los tests de infección latente por tuberculosis pueden ser falsamente negativos. El riesgo de resultados de tests cutáneos de tuberculina falsos negativos se debe considerar especialmente en pacientes que están severamente enfermos o inmunocomprometidos. Se debe indicar a los pacientes que acudan al médico si advierten signos o síntomas (p. ej., tos persistente, debilidad/pérdida de peso, bajo grado de fiebre) sugerentes de que puede ocurrir una infección por tuberculosis. Otras Infecciones Oportunísticas: Infecciones oportunísticas, incluyendo infecciones invasivas por hongos, se han observado en pacientes que reciben Humira. Estas infecciones no se reconocen consistentemente en pacientes que toman bloqueadores anti-TNF y esto ha producido retrasos en un tratamiento apropiado, algunas veces produciendo resultados fatales. Los pacientes que toman bloqueadores anti-TNF son más susceptibles para infecciones serias por hongos tales como histoplasmosis, coccidioidomicosis, blastomicosis, aspergillosis, candidiasis, y otras infecciones oportunísticas. Aquellos que desarrollan fiebre, malestar, pérdida de peso, sudoración, tos, disnea, y/o infiltrados pulmonares, u otras enfermedades sistémicas serias con o sin shock concomitante deben buscar atención médica de inmediato para una evaluación diagnóstica. Para los pacientes que residen o viajan en regiones donde las micosis son endémicas, se deben sospechar infecciones invasivas por hongos si ellos desarrollan signos y síntomas de posible infección sistémica por hongos. Los pacientes están en alto riesgo de histoplasmosis y otras infecciones invasivas por hongos y por consiguiente los clínicos deben considerar tratamiento empírico antihongos hasta que el patógeno(s) sea identificado. El test para antígeno y anticuerpo para histoplasmosis puede ser negativo en algunos pacientes con infección activa. Cuando sea posible, la decisión de administrar terapia antifúngica empírica en estos pacientes se debe hacer consultando a un médico con experiencia en el diagnóstico y tratamiento de infecciones invasivas por hongos y se debe tener en consideración tanto el riesgo de infección severa por hongos como los riesgos de la terapia antifúngica. Los pacientes que desarrollan una infección severa por hongos también deben ser aconsejados para suspender el bloqueador de TNF hasta que las infecciones se controlen. Reactivación de Hepatitis B: El uso de bloqueadores TNF se ha asociado con la reactivación del virus de la hepatitis B (VHB) en pacientes que son portadores crónicos de este virus. En algunos casos, la reactivación del VHB que ocurre en conjunto con la terapia con bloqueador TNF ha sido fatal. La mayoría de estos informes ha ocurrido en pacientes que concomitante recibían otras drogas que suprimen el sistema inmune, que también pueden contribuir a la reactivación del VHB. Los pacientes con riesgo de infección por VHB deben ser evaluados por evidencia previa de infección por VHB antes de iniciar terapia con bloqueador TNF. Los médicos deben tener precaución al prescribir bloqueadores TNF en pacientes identificados como portadores del VHB. Los pacientes que son portadores del VHB y requieren tratamiento con bloqueadores TNF deben ser vigilados de cerca para signos y síntomas de infección activa por VHB a través de la terapia y por varios meses después del término de la terapia. No existen datos disponibles adecuados en relación con la seguridad o eficacia en pacientes tratados que son portadores del VHB con terapia antiviral junto con la terapia con bloqueador TNF para prevenir la reactivación del VHB. En los pacientes que desarrollan una reactivación del VHB, Humira se debe discontinuar y se debe iniciar una terapia antiviral efectiva con tratamiento de apoyo apropiado. Eventos neurológicos: Los antagonistas del TNF, incluido Humira, se han asociado a casos raros de nuevos episodios o exacerbación de síntomas clínicos y/o evidencia radiográfica de enfermedad desmielinizante, incluyendo esclerosis múltiple. Los médicos deben tener precaución al considerar el uso de Humira en pacientes con trastornos desmielinizantes del sistema nervioso central preexistentes o de inicio reciente. Neoplasias Malignas: En los brazos controlados de estudios clínicos con antagonistas-TNF, se han observado más casos de neoplasias malignas incluyendo linfoma entre los pacientes que recibían un antagonista-TNF comparado con los pacientes control. El tamaño del grupo control y la duración limitada de los brazos controlados de los estudios imposibilita la capacidad de dar conclusiones firmes. Además, existe un antecedente de aumento del riesgo de linfoma en pacientes con artritis reumatoidea con larga data de enfermedad, altamente activa, enfermedad inflamatoria, que complica la estimación del riesgo. Durante los estudios abiertos a largo plazo con Humira, la tasa total de neoplasias malignas fue similar a la que se esperaría para la edad, sexo y raza cruzada con la población general. Con el conocimiento actual, no se puede excluir un posible riesgo de desarrollo de linfomas u otras neoplasias malignas en los pacientes tratados con un antagonista-TNF. Neoplasias malignas, algunas fatales, se han reportado entre niños y adolescentes (inicio de la terapia a los 18 años de edad o menos) que recibieron tratamiento con agentes bloqueadores del TNF. Aproximadamente la mitad de los casos fueron linfomas, incluyendo linfoma Hodgkin y no-Hodgkin. Los otros casos representaron una variedad de diferentes neoplasias malignas e incluyeron neoplasias malignas raras generalmente asociadas con inmunosupresión y neoplasias no observadas habitualmente en niños y adolescentes. Las neoplasias malignas ocurrieron después de una mediana de 30 meses de terapia. La mayoría de los pacientes estaban recibiendo inmunosupresores concomitantes. Estos casos fueron reportados post-comercialización y fueron derivados de una variedad de fuentes incluyendo registros y reportes espontáneos post-comercialización. Reportes post-marketing muy raro de linfoma de células T hepatoesplénico (HSTCL), un raro linfoma agresivo que es frecuentemente fatal, se han identificado en pacientes tratados con adalimumab. La mayoría de los pacientes tuvo terapia previa con infliximab así como uso concomitante con azatioprina o 6-mercaptopurina para enfermedad de Crohn. La asociación causal de HSTCL con adalimumab no es clara. No se han realizado estudios que incluyan pacientes con una historia de neoplasia maligna o que continúan en tratamiento pacientes que desarrollaron neoplasia maligna mientras recibían Humira. Por lo tanto, se debe tener precaución adicional en la consideración de tratamiento con Humira de estos pacientes. Todos los pacientes, y en particular pacientes con una historia médica de extensa terapia inmunosupresora o pacientes con psoriasis con una historia de tratamiento con PUVA se deben examinar por la presencia de cáncer de piel no-melanoma previo a y durante el tratamiento con Humira. Se han reportado casos de leucemia aguda y crónica en asociación con el uso de bloqueador anti-TNF post-comercialización en artritis reumatoidea y otras indicaciones. Los pacientes con artritis reumatoidea pueden tener un mayor riesgo (hasta dos veces) que la población general para el desarrollo de leucemia, aún en ausencia de terapia con bloqueadores anti-TNF. Alergias: Las reacciones alérgicas serias asociadas con Humira fueron raras durante los estudios clínicos. En la post comercialización, reacciones alérgicas serias incluyendo anafilaxis se han informado muy raramente después de la administración de Humira. Si se produce una reacción anafiláctica u otra reacción alérgica seria, se debe suspender inmediatamente la administración de Humira e iniciar un tratamiento apropiado. La cubierta de la aguja de la jeringa contiene caucho natural (látex). Esto puede causar reacciones alérgicas severas en pacientes sensibles al látex. Eventos Hematológicos: Se han informado raros reportes de pancitopenia incluyendo anemia aplástica con agentes bloqueadores del TNF. Eventos adversos del sistema hematológico, incluyendo citopenia médicamente significativa (e.j. trombocitopenia, leucopenia) se han informado con Humira. La relación causal de estos informes de Humira sigue siendo confusa. Todos los pacientes mientras están en tratamiento con Humira deben ser aconsejados de buscar atención médica inmediata si desarrollan signos y síntomas sugerentes de discrasias sanguíneas (e.j. fiebre persistente, hematomas, sangrados, palidez). La discontinuación de la terapia con Humira se debe considerar en pacientes con anormalidades hematológicas significativas confirmadas. Uso con Anakinra: Infecciones serias se han observado en estudios clínicos con el uso concurrente de anakinra y otro antagonista-TNF, etanercept, sin que agregue beneficios clínicos comparado con etanercept solo. Debido a la naturaleza de los eventos adversos observados con la combinación de la terapia con etanercept y anakinra, se pueden producir también toxicidades similares con la combinación de anakinra y otros antagonistas-TNF. Por lo tanto, la combinación de adalimumab y anakinra no se recomienda. Uso con Abatacept: La administración concurrente de antagonistas-TNF y abatacept se ha asociado con un aumento del riesgo de infecciones incluyendo infecciones serias comparado con antagonistas-TNF solos. Esta combinación no ha demostrado aumento del beneficio clínico. Por lo tanto, el uso combinado de antagonistas-TNF y abatacept no se recomienda. Inmunosupresión: En un estudio de 64 pacientes con AR que recibieron tratamiento con Humira, no se observó evidencia de depresión de la hipersensibilidad de tipo retardado, depresión de los niveles de inmunoglobulina ni cambios en los recuentos de células-B y células-T efectoras, y células-NK, monocitos/macrófagos y neutrófilos. Vacunas: En un estudio randomizado, doble ciego, placebo controlado en 226 pacientes adultos con artritis reumatoidea tratados con Humira, se evaluaron las respuestas de anticuerpos a las vacunas pneumocócica e influenza. Los niveles de anticuerpos protectores a los antígenos pneumocócicos se alcanzaron en 86% de los pacientes en el grupo de Humira comparado a 82% en el grupo placebo. Un total de 37% de los sujetos tratados con Humira y 40% de los sujetos tratados con placebo alcanzaron al menos un aumento de 2 veces en al menos 3 de 5 antígenos pneumocócicos. En el mismo estudio 98% de los pacientes en el grupo Humira y 95% en el grupo placebo alcanzaron niveles protectores de anticuerpos a los antígenos de influenza. Un total de 52% de los sujetos tratados con Humira y 63% de los sujetos tratados con placebo alcanzaron al menos un aumento de 4 veces en al menos 2 de 3 antígenos de influenza. Se recomienda que los pacientes con artritis idiopática juvenil poliarticular, si es posible, estén al día con todas las inmunizaciones de acuerdo con las actuales guías de inmunización previo al inicio de la terapia con Humira. Los pacientes con Humira pueden recibir vacunas concurrentes, excepto vacunas vivas. No existen datos disponibles sobre la transmisión secundaria de infección por vacunas vivas en pacientes que reciben Humira. Insuficiencia Cardíaca Congestiva: Humira no se ha estudiado formalmente en pacientes con insuficiencia cardíaca congestiva (ICC), sin embargo, en estudios clínicos con otro antagonista-TNF, se han informado una mayor tasa de eventos adversos serios relacionados con ICC incluyendo agravamiento de ICC e inicio de nuevos cuadros de ICC. También se han reportado casos de agravamiento de ICC en pacientes que recibían Humira. Los médicos deben extremar las precauciones al usar Humira en pacientes que tienen insuficiencia cardíaca y supervisarlos cuidadosamente. Procesos Autoinmunes: El tratamiento con Humira puede producir formación de anticuerpos autoinmunes. El impacto del tratamiento a largo plazo con Humira en el desarrollo de enfermedades autoinmunes no se conoce. Si un paciente desarrolla síntomas sugerentes de un síndrome tipo lupus después de tratamiento con Humira, el tratamiento se debe discontinuar (ver Reacciones adversas - Autoanti
cuerpos). Uso Geriátrico: La frecuencia de infección seria entre los sujetos sobre 65 años de edad tratados con Humira fue mayor que para aquellos bajo 65 años de edad. Del número total de sujetos en estudios clínicos de Humira, 12% tenían 65 años y más, mientras que aproximadamente 2.5% tenían 75 años y más. Debido a que existe una mayor incidencia de infecciones en los adultos mayores en la población general, se debe tener precaución cuando se trata a adultos mayores.
Interacciones.
Cuando Humira se administra a 21 pacientes con AR con terapia estable con MTX, no hubo cambios estadísticamente significativos en los perfiles de concentración séricos de MTX. En contraste, después de dosis únicas y múltiples, MTX redujo los clearances aparentes de adalimumab en 29% y 44% respectivamente. Los datos no sugieren la necesidad de ajuste de dosis de ya sea Humira o MTX. No se han evaluado las interacciones entre Humira y fármacos distintos de MTX en estudios farmacocinéticos formales. En estudios clínicos, no se han observado interacciones cuando Humira se administró con DMARDS de uso habitual (sulfasalazina, hidrocloroquina, leflunomida y oro parenteral), glucocorticoides, salicilatos, antiinflamatorios no esteroidales o analgésicos. Interacción entre el Fármaco y Análisis de Laboratorio: No existen interferencias conocidas entre Humira y análisis de laboratorio.
Conservación.
Conservar entre 2°C a 8°C (36°F - 46°F) (en un refrigerador) y conservar la jeringa o vial en el envase de cartón externo. No congelar. No utilizar después de la fecha de vencimiento.
Sobredosificación.
No se ha establecido la dosis máxima tolerada de Humira en seres humanos. No se han observado toxicidades limitantes de la dosis durante los estudios clínicos de HUMIRA. Se han administrado múltiples dosis de hasta 10 mg/kg a pacientes en estudios clínicos sin evidencia de toxicidades limitantes de dosis. En caso de sobredosis, se recomienda vigilar en el paciente la aparición de signos o síntomas de efectos o reacciones adversas e instaurar inmediatamente el tratamiento sintomático apropiado.
Presentación.
Humira (adalimumab) solución inyectable se presenta en forma de solución estéril de 40 mg de adalimumab disueltos en 0.8 mL de solución estéril para administración parenteral en las siguientes opciones de envasado: Humira 40 mg solución inyectable en jeringa precargada para un solo uso (para uso por el paciente): Envase de cartón que contiene 2 blisters con 1 jeringa precargada y 1 gasa con alcohol en cada blister. Humira 40 mg solución inyectable en un Pen (dispositivo de aplicación - autoinyector) precargado: Envase de cartón que contiene dos gasas preparadas con alcohol y dos blisters de dosis. Cada blister de dosis consiste de un Pen de uso único, que contiene una jeringa de vidrio prellenada con 1 mL, con una aguja fija calibre 27½ pulgadas, proporcionando 40 mg (0.8 mL) de Humira.