LUVERIS 75UI
MERCK
Gonadotrofinoterapia.
Composición.
Cada vial com liofilizado de Luveris® 75 UI contiene Lutropina alfa (r-hLH) 75 UI. Excipientes: sacarosa, fosfato disódico dihidratado, fosfato dihidrogeno sódico monohidratado, polisorbato 20, metionina, hidróxido de sódio (para ajuste de ph), ácido fosfórico concentrado (para ajuste de pH). Cada vial con solvente contiene: Agua para inyectable 1 mL. Forma farmacéutica: Polvo y disolvente para solución inyectable.
Indicaciones.
Luveris®, asociado a un preparado de hormona foliculoestimulante (FSH), está recomendado para la estimulación del desarrollo folicular en mujeres adultas con déficit severo de Hormona Luteinizante (LH) y FSH. En los ensayos clínicos, estas pacientes se definieron por un nivel sérico de LH endógena de < 1,2 U.I./L.
Dosificación.
El tratamiento con Luveris® debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de los trastornos de fertilidad. Posología: En mujeres con déficit de LH y FSH, el objetivo del tratamiento con Luveris® asociado a FSH es desarrollar un único folículo de Graaf maduro, a partir del cual se liberará el ovocito tras la administración de gonadotropina coriónica humana (hCG). Luveris® debe administrarse como un ciclo de inyecciones diarias, conjuntamente con FSH. Puesto que estas pacientes son amenorreicas y tienen una escasa secreción endógena de estrógenos, el tratamiento puede comenzar en cualquier momento. Luveris® se administra de forma concomitante con folitropina alfa. El tratamiento debe adaptarse a la respuesta individual de la paciente, evaluada mediante el tamaño folicular determinado por una ecografía y la respuesta estrogénica. Una pauta recomendada comienza con 75 UI de lutropina alfa (es decir, un vial de Luveris®) por día y 75-150 UI de FSH. Los ensayos clínicos han demostrado que Luveris® aumenta la sensibilidad ovárica a la folitropina alfa. Si se considera adecuado aumentar la dosis de FSH, se debe hacer preferentemente a intervalos de 7-14 días y preferentemente con incrementos de 37,5-75 UI. Puede ser aceptable que la duración de la estimulación en un ciclo determinado se prolongue hasta 5 semanas. Cuando se obtiene una respuesta óptima, debe administrarse una inyección única de 250 microgramos de r-hCG o de 5.000 UI a 10.000 UI de hCG, 24-48 horas después de la última inyección de Luveris® y de FSH. Se recomienda a la paciente que realice el coito el mismo día de la administración de hCG, así como al día siguiente. De forma alternativa, se puede realizar inseminación intrauterina (IIU). Puede ser preciso el apoyo de la fase lútea, ya que la falta de sustancias con actividad luteotropa (LH/hCG) después de la ovulación puede dar lugar a un fracaso prematuro del cuerpo lúteo. Si se obtiene una respuesta excesiva, debe interrumpirse el tratamiento y no administrarse hCG. El tratamiento debe reiniciarse en el ciclo siguiente con una dosis de FSH más baja que la del ciclo previo. Forma de administración: Luveris® se administra por vía subcutánea. El sitio de inyección se debe alternar diariamente. El polvo debe ser reconstituido justo antes de usarlo, con el disolvente suministrado. La autoadministración de este medicamento sólo debe realizarse por pacientes adecuadamente motivadas y entrenadas para ello, con acceso al consejo de un profesional.
Contraindicaciones.
Luveris® está contraindicado en pacientes con: hipersensibilidad a la lutropina alfa o a alguno de los excipientes; tumores del hipotálamo o de la hipófisis; aumento del tamaño de los ovarios o la presencia de quistes ováricos de origen desconocido; hemorragias ginecológicas de origen desconocido. Carcinoma ovárico, uterino o de mama.
Embarazo y lactancia.
Embarazo: No hay indicaciones de uso de Luveris® durante el embarazo. Los datos de un número limitado de embarazos expuestos indican que no hay reacciones adversas de las gonadotropinas durante el embarazo, el desarrollo embrionario o fetal, el parto o el desarrollo postnatal tras una estimulación ovárica controlada. No se han observado efectos teratógenos de Luveris® en estudios en animales. En caso de exposición durante el embarazo, los datos clínicos no son suficientes como para descartar que Luveris® tenga efectos teratógenos. Lactancia: Luveris® no está indicado durante la lactancia. Durante la misma, la secreción de prolactina puede llevar a un mal pronóstico de la estimulación ovárica.
Reacciones adversas.
Para la terminología de frecuencia de las reacciones adversas se utilizarán las siguientes definiciones de aquí en adelante: Muy frecuentes (≥1/10). Frecuentes (≥1/100 a < 1/10). Poco frecuentes (≥1/1.000 a < 1/100). Raras (≥1/10.000 a < 1/1.000). Muy raras ( < 1/10.000). Frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Pueden observarse las siguientes reacciones adversas tras la administración de Luveris®. Trastornos del sistema inmunológico: Muy raras: reacciones de hipersensibilidad de leves a graves, incluyendo shock y reacciones anafilácticas. Trastornos del sistema nervioso: Frecuentes: cefalea. Trastornos vasculares: Muy raras: tromboembolismo, asociado por lo general a SHO grave. Trastornos gastrointestinales: Frecuentes: dolor abdominal, molestias abdominales, náuseas, vómitos, diarrea. Trastornos del aparato reproductor y de la mama: Frecuentes: SHO leve o moderado (incluidos los síntomas asociados), quiste ovárico, dolor de mama, dolor pélvico. Trastornos generales y alteraciones en el lugar de administración: Frecuentes: reacción en el lugar de inyección (p. ej., dolor, eritema, hematoma, inflamación y/o irritación en el lugar de inyección).
Advertencias.
Antes de iniciar el tratamiento, debe valorarse adecuadamente el tipo de infertilidad de la pareja y la posible existencia de contraindicaciones para el embarazo. Además, debe descartarse la presencia de hipotiroidismo, insuficiencia suprarrenal e hiperprolactinemia, instaurando el tratamiento específico apropiado. En pacientes con porfiria o antecedentes de porfiria, Luveris® puede aumentar el riesgo de un ataque agudo. Si se produce un empeoramiento de esta afección o si esta se manifiesta por primera vez, puede ser necesario interrumpir el tratamiento. Síndrome de hiperestimulación ovárica (SHO): Uno de los posibles efectos de la estimulación ovárica controlada es un cierto grado de aumento de tamaño de los ovarios. Se suele presentar con mayor frecuencia en mujeres con síndrome del ovario poliquístico y por lo general remite sin tratamiento. A diferencia de un aumento de tamaño de los ovarios sin complicaciones, el SHO es una afección que se manifiesta con grados crecientes de gravedad. Comprende un notable aumento de tamaño de los ovarios, una gran cantidad en suero de esteroides sexuales y un aumento de la permeabilidad vascular que puede provocar una acumulación de líquido en las cavidades peritoneal, pleural y, en raras ocasiones, pericárdica. Entre las manifestaciones leves del SHO se pueden citar dolor abdominal, molestias abdominales y distensión o aumento de tamaño de los ovarios. Un SHO moderado puede adicionalmente presentar náuseas, vómitos, hallazgo de ascitis en ecografías o aumento notable de tamaño de los ovarios. Un SHO grave adicionalmente incluye síntomas como un gran aumento del tamaño de los ovarios, aumento de peso, disnea u oliguria. En una exploración clínica se pueden hallar signos tales como hipovolemia, hemoconcentración, desequilibrios electrolíticos, ascitis, derrames pleurales o dificultad respiratoria aguda. En casos muy raros, el SHO grave puede complicarse con torsión ovárica o episodios tromboembólicos como pueden ser embolia pulmonar, ictus isquémico o infarto de miocardio. Entre los factores de riesgo independientes de aparición de SHO se pueden citar edad joven, masa corporal escasa, síndrome del ovario poliquístico, dosis elevadas de gonadotropinas exógenas, niveles absolutos elevados o en rápido ascenso de estradiol sérico y episodios anteriores de SHO, gran número de folículos ováricos en desarrollo y gran número de ovocitos recuperados en los ciclos de TRA. El riesgo de hiperestimulación ovárica puede minimizarse utilizando la posología y el esquema posológico de Luveris® y FSH recomendados. También se recomienda monitorizar cuidadosamente los ciclos de estimulación por medio de ecografías así como también los niveles de estradiol para así identificar precozmente cualquier factor de riesgo. Se han encontrado indicios de que la hCG desempeña un papel fundamental en la aparición del SHO y de que el síndrome puede ser más grave y tener una duración mayor si se produce un embarazo. Por tanto, en caso de que se manifiesten signos de hiperestimulación ovárica, se recomienda no administrar hCG y advertir a la paciente que no realice el coito o que utilice métodos anticonceptivos de barrera durante al menos 4 días. Dado que el SHO puede evolucionar con gran rapidez (en 24 horas) o a lo largo de varios días hasta convertirse en una situación médica grave, las pacientes deberán estar sometidas a seguimiento durante al menos dos semanas tras la administración de hCG. El SHO leve o moderado remite por lo general de modo espontáneo. En caso de SHO grave, se recomienda interrumpir el tratamiento con gonadotropinas (si en ese momento se está administrando) y hospitalizar a la paciente e iniciar el tratamiento apropiado. Embarazo múltiple: En pacientes sometidas a la inducción de la ovulación, la incidencia de embarazo múltiple es más elevada que en el caso de concepción natural. La mayor parte de los embarazos múltiples son gemelares. Los embarazos múltiples, en especial aquellos con un número elevado de fetos, conllevan un riesgo mayor de que el desenlace clínico materno y perinatal sea adverso. Para minimizar el riesgo de embarazo múltiple de mayor orden, se recomienda monitorizar cuidadosamente la respuesta ovárica. En pacientes sometidas a Técnicas de Reproducción Asistida (TRA) el riesgo de embarazo múltiple está asociado principalmente al número de embriones transferidos, su calidad y la edad de la paciente. Pérdida del embarazo: La incidencia de pérdida del embarazo por aborto espontáneo o provocado es mayor en pacientes sometidas a estimulación del crecimiento folicular para inducción de la ovulación que con la concepción natural. Embarazo ectópico: Las mujeres con antecedentes de enfermedad tubárica tienen riesgo de embarazo ectópico, independientemente de que el embarazo se consiga mediante concepción espontánea o con tratamientos de fertilidad. Se ha notificado que la prevalencia de embarazo ectópico tras practicar TRA es superior a la de la población general. Malformaciones congénitas: La prevalencia de malformaciones congénitas tras el uso de TRA puede ser ligeramente superior que tras embarazos espontáneos. Ello podría deberse a factores relacionados con los progenitores (por ejemplo, edad de la madre, genética), intervenciones relacionadas con TRA y embarazos múltiples. Episodios tromboembólicos: En mujeres que hayan sufrido recientemente o estén sufriendo una afección tromboembólica o en mujeres con factores de riesgo generalmente reconocidos de tromboembolia, como antecedentes personales o familiares, trombofilia u obesidad grave (índice de masa corporal > 30 kg/m2), el tratamiento con gonadotropinas puede aumentar aún más el riesgo de empeoramiento o aparición de este tipo de episodios. En estas mujeres, deben sopesarse los beneficios y los riesgos de la administración de gonadotropinas. No obstante, conviene señalar que el embarazo en sí mismo, así como el SHO, también entraña un mayor riesgo de tromboembolia. Neoplasias del aparato reproductor: Se han notificado neoplasias de ovario y de otras partes del aparato reproductor, tanto benignas como malignas, en mujeres que han recibido diversos tratamientos farmacológicos para la infertilidad. Aún está por determinar si el tratamiento con gonadotropinas aumenta el riesgo de estos tumores en mujeres no fértiles. Poblaciones especiales: Población pediátrica: No existen indicaciones relevantes para el uso de Luveris® en la población pediátrica. Pacientes de edad avanzada: No existen indicaciones relevantes para el uso de Luveris® en pacientes de edad avanzada. No se ha establecido la seguridad y eficacia de Luveris® en pacientes de edad avanzada. Pacientes con insuficiencia renal o hepática: No se ha establecido la seguridad, la eficacia y la farmacocinética de Luveris® en pacientes con insuficiencia renal o hepática. Efectos sobre la capacidad de conducir o utilizar máquinas: No se han realizado estudios sobre los efectos de Luveris® sobre la capacidad para conducir y usar máquinas.
Interacciones.
Luveris® no debe mezclarse con otros medicamentos en la misma jeringa, excepto con folitropina alfa, ya que los estudios correspondientes han demostrado que la coadministración de ambos fármacos no altera significativamente la actividad, estabilidad, ni las propiedades farmacocinéticas o farmacodinámicas de los principios activos. No ha sido reportada durante la terapia con Luveris® ninguna otra interacción con medicamentos que sea clínicamente significante.
Conservación.
No conservar a temperatura superior a 25°C. Conservar en el embalaje original para protegerlo de la luz. No utilice Luveris® si observa indicios visibles de deterioro, tales como decoloración del polvo o daño del envase. El medicamento debe administrarse inmediatamente después de disolver el polvo. La solución no debe administrarse si contiene partículas o no es límpida. Advertencias de este y todos los medicamentos: Mantener fuera del alcance de los niños. Úsese sólo por indicación y bajo supervisión médica. No repita el medicamento sin indicación del médico. No utilice este medicamento si observa signos visibles de deterioro. No use medicamentos vencidos. En caso de sobredosis concurra al Centro Asistencial más próximo. Instrucciones de uso: Luveris® debe administrarse por vía subcutánea, es decir, poniendo la inyección bajo la piel. Cada vial es para un solo uso. Si se administra Luveris® usted misma, lea con detenimiento las siguientes instrucciones: Lávese las manos. Es importante que sus manos y los materiales que utilice estén lo más limpios posible. Reúna todo lo que vaya a necesitar. Busque un lugar limpio y prepárelo todo: un vial de Luveris®, un vial de disolvente, dos torundas de algodón empapadas en alcohol, una jeringa, una aguja de reconstitución para disolver el polvo con el disolvente, una aguja fina para la inyección subcutánea, un recipiente para objetos cortantes para desechar con precaución los envases de vidrio y las agujas. Quite la cápsula de cierre protectora del vial de disolvente: Coloque la aguja de reconstitución en la jeringa e introduzca algo de aire en la jeringa tirando del émbolo aproximadamente hasta la marca de 1 mL. A continuación, introduzca la aguja en el vial, empuje el émbolo para expulsar el aire, coloque el vial boca abajo y extraiga suavemente todo el disolvente.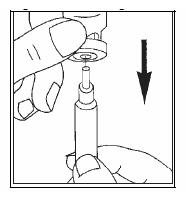
Ponga la jeringa con cuidado sobre la mesa, procurando no tocar la aguja. Prepare la solución para inyección: Retire la cápsula de cierre protectora del vial que contiene el polvo de Luveris®, coja su jeringa e inyecte lentamente el disolvente en el vial de Luveris®. Muévalo suavemente sin retirar la jeringa. No lo agite.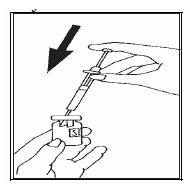
Una vez que el polvo se haya disuelto (lo que suele ocurrir inmediatamente), compruebe que la disolución obtenida es límpida y no contiene ninguna partícula. Ponga el vial boca abajo y cargue suavemente la solución en la jeringa. Usted también puede mezclar Luveris® y folitropina alfa, como alternativa a inyectar cada producto por separado. Después de disolver el polvo de Luveris®, cargue la solución en la jeringa y vuelva a inyectarla en el envase que contenga el polvo de folitropina alfa. Una vez que se haya disuelto el polvo, extraiga la solución con la jeringa. Compruebe si existen partículas, como se indica más arriba, y no utilice la solución si no es límpida. Se pueden reconstituir hasta 3 envases de polvo en 1 mL de disolvente. Cambie la aguja, colocando la aguja fina y elimine las posibles burbujas de aire: Si ve alguna burbuja de aire en la jeringa, tome ésta con la aguja hacia arriba y dé golpecitos en la jeringa hasta que el aire se reúna en la parte superior. Empuje el émbolo suavemente hasta que desaparezcan las burbujas de aire.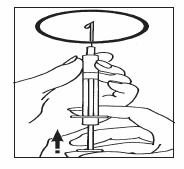
Inyecte la solución inmediatamente: Su médico o enfermero le habrán indicado dónde debe poner la inyección (p. ej., vientre, parte delantera del muslo). Limpie la zona elegida con un algodón embebido de alcohol. Pellizque enérgicamente la piel e introduzca la aguja con un ángulo de 45 a 90°, con un movimiento similar al de los dardos. Inyecte bajo la piel, según las instrucciones recibidas. No inyecte directamente en una vena. Introduzca la solución presionando suavemente sobre el émbolo. Emplee todo el tiempo que necesite hasta inyectar la totalidad de la solución. Retire inmediatamente la aguja y limpie la piel con un algodón embebido de alcohol realizando un movimiento circular.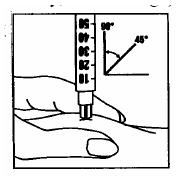
Deseche todo el material: Una vez finalizada la inyección, deseche inmediatamente todas las agujas y envases de vidrio vacíos en el recipiente para objetos cortantes que se suministra. Debe desecharse cualquier porción de la solución no utilizada.
Sobredosificación.
Los efectos de una sobredosis de Luveris® son desconocidos. Sin embargo, puede esperarse que se produzca un SHO. Se han administrado dosis únicas de hasta 40.000 UI de lutropina alfa en voluntarias sanas sin que se observaran reacciones adversas graves y con una buena tolerancia. Atención terapéutica: El tratamiento irá dirigido a los síntomas.
Presentación.
Cada envase contiene 1 vial con liofilizado para solución inyectable.