MIRCERA®
ROCHE
MIRCERA es la primera molécula de un nuevo grupo de activadores continuos del receptor de la eritropoyetina; su nombre es metoxipolietilenglicol-epoetina beta.
Composición.
Jeringas precargadas monodosis: con 50 mg, 75 mg, 100 mg, 150 mg, 200 mg o 250 mg de metoxipolietilenglicol-epoetina beta en 0,3 ml u 800 de metoxipolietilenglicol-epoetina beta en 0,6mg ml. El principio activo, metoxipolietilenglicol-epoetina beta, es un conjugado covalente de una proteína obtenida por técnicas de ADN recombinante en células de ovario de hámster chino y de un metoxi-polietilenglicol (PEG) lineal. Su peso molecular es de aproximadamente 60 kDa. La concentración de la dosis expresada en mg indica la cantidad de fracción proteica de la molécula de metoxipolietilenglicol-epoetina beta, sin considerar la porción glucosilada. La solución es límpida e incolora o ligeramente amarillenta. Excipientes: fosfato de sodio monobásico monohidrato, sulfato de sodio, manitol, metionina, poloxámero 188, ácido clorhídrico (diluido), solución de hidróxido de sodio y agua para inyectables. Forma farmacéutica: Solución para inyección estéril lista para el uso, presentada en: jeringas precargadas monodosis. Vía de administración: Subcutánea o intravenosa. Declaración de esterilidad / radiactividad: No procede.
Farmacología.
Propiedades farmacodinámicas: MIRCERA es un activador continuo del receptor de la eritropoyetina, obtenido por síntesis química. La metoxipolietilenglicol-epoetina beta difiere de la eritropoyetina por la inserción de un enlace amídico entre el grupo amino N-terminal o el grupo amino en el carbono e de la lisina, sobre todo en los residuos de las posiciones 52 (Lys52) y 45 (Lys45), y el ácido metoxipolietilenglicolbutanoico. Como consecuencia de ello, el peso molecular de la metoxipolietilenglicol-epoetina beta es de aproximadamente 60.000 Da, y el de la porción PEG, de aproximadamente 30.000 Da. La actividad de MIRCERA en los receptores es diferente a la de la eritropoyetina, y se caracteriza por una asociación más lenta y una disociación más rápida con los receptores, una actividad específica reducida in vitro con una actividad aumentada in vivo, así como una semivida más larga. Estas propiedades farmacológicas diferenciales son importantes para la pauta de administración de una dosis mensual de MIRCERA. Mecanismo de acción: MIRCERA estimula la eritropoyesis mediante su interacción con el receptor de la eritropoyetina en las células progenitoras de la médula ósea. Como factor de crecimiento primario para el desarrollo eritroide, la hormona natural eritropoyetina se produce en los riñones y se secreta a la corriente sanguínea en presencia de un estado hipóxico. En su respuesta a la hipoxia, la hormona natural eritropoyetina interactúa con las células progenitoras eritroides para incrementar la producción de glóbulos rojos. Estudios clínicos / Eficacia: En dos estudios aleatorizados y controlados en pacientes con IRC no dializados, BA16738 y NH20052, se alcanzó la corrección de la anemia con MIRCERA en el 97,5% y el 94,1% de los pacientes, respectivamente. En las ocho primeras semanas de tratamiento, la proporción de pacientes con una concentración de hemoglobina superior a 13 g/dl fue del 11,4% en el grupo de MIRCERA y del 34% en de darbepoetina alfa en el estudio BA16738, mientras que las proporciones correspondientes de pacientes con una cifra de hemoglobina superior a 12 g/dl fueron del 25,8% en el grupo de MIRCERA y del 47,7% en el de darbepoetina alfa en el estudio NH20052. En un estudio aleatorizado y controlado en pacientes con IRC dializados se alcanzó la corrección de la anemia con MIRCERA en el 93,3% de los pacientes. Se realizaron cuatro estudios aleatorizados y controlados en pacientes dializados y tratados a la vez con darbepoetina alfa o epoetina. Se distribuyó aleatoriamente a los pacientes entre un grupo de mantenimiento del tratamiento y otro de cambio a MIRCERA para alcanzar una concentración estable de hemoglobina. En el período de evaluación (semanas 29 a 36), los valores medio y mediano de hemoglobina en los pacientes tratados con MIRCERA eran prácticamente idénticos a los valores basales. Propiedades farmacocinéticas: Con su larga semivida de eliminación, las propiedades farmacocinéticas y farmacológicas de MIRCERA permiten su administración una vez al mes. La semivida de eliminación de MIRCERA tras su administración i.v. es de 15 a 20 veces más larga que la de la eritropoyetina recombinante humana. La farmacocinética de MIRCERA se ha estudiado en voluntarios sanos y en pacientes anémicos con IRC, incluidos pacientes dializados y no dializados. En los pacientes con IRC, el aclaramiento y el volumen de distribución de la metoxipolietilenglicol-epoetina beta no dependían de la dosis. En los pacientes con IRC, la farmacocinética de MIRCERA se evaluó después de la primera dosis y tras su administración en la semana 9 y en la semana 19 o 21. Las dosis múltiples no tenían ningún efecto en el aclaramiento, el volumen de distribución y la biodisponibilidad de la metoxipolietilenglicol-epoetina beta. Después de la administración cada 4 semanas a pacientes con IRC no se detectó una acumulación significativa de metoxipolietilenglicol-epoetina beta (razón de acumulación: 1,03). La comparación de las concentraciones séricas de metoxipolietilenglicol-epoetina beta determinadas antes y después de la hemodiálisis en 41 pacientes con IRC puso de manifiesto que la hemodiálisis no tenía ningún efecto en la farmacocinética de la metoxipolietilenglicol- epoetina beta. Un análisis en 126 pacientes con IRC no mostró ninguna diferencia entre los pacientes dializados y los no dializados. Los resultados de un estudio en 42 voluntarios sanos revelaron que el sitio de la inyección subcutánea (abdomen, brazo o muslo) no influía de manera clínicamente relevante en la farmacocinética, la farmacodinámica y la tolerabilidad local de MIRCERA. A la vista de estos resultados, los tres sitios se consideran adecuados para la inyección subcutánea de MIRCERA. Absorción: Absorción tras la administración subcutánea: Tras la administración subcutánea a pacientes con IRC no dializados, la concentración sérica máxima de metoxipolietilenglicol-epoetina beta se alcanzó al cabo de 95 horas (mediana). La biodisponibilidad absoluta de la metoxipolietilenglicol-epoetina beta tras la administración s.c. fue del 54%. La semivida de eliminación terminal en los pacientes con IRC no dializados fue de 142 horas. Tras la administración subcutánea a pacientes con IRC dializados, la concentración sérica máxima de metoxipolietilenglicol-epoetina beta se alcanzó al cabo de 72 horas (mediana). La biodisponibilidad absoluta de la metoxipolietilenglicol-epoetina beta tras la administración s.c. fue del 62%. La semivida de eliminación terminal en los pacientes con IRC dializados fue de 139 horas. Distribución: Un estudio en 400 pacientes con IRC ha mostrado que el volumen de distribución de la metoxipolietilenglicol-epoetina beta es de aproximadamente 5 litros. Eliminación: Tras la administración i.v. a pacientes con IRC, la semivida de eliminación terminal (t1/2) de la metoxipolietilenglicol-epoetina beta era de 134 horas (o 5,6 días), y el aclaramiento sistémico total, de 0,494 ml/h por kg. Después de la administración s.c., t1/2 era de 139 horas en los pacientes dializados y de 142 horas en los no dializados. Farmacocinética en poblaciones especiales: Insuficiencia hepática: La farmacocinética de MIRCERA en pacientes con insuficiencia hepática es similar a la de los sujetos sanos (ver Pautas posológicas especiales). Otras poblaciones especiales: Mediante análisis poblacionales se han evaluado los efectos potenciales de las características demográficas en la farmacocinética de MIRCERA. Los resultados de estos análisis ponen de manifiesto que no es necesario ajustar la dosis inicial en función de la edad, el sexo o la raza. Un análisis poblacional de farmacocinética tampoco reveló diferencias farmacocinéticas entre los pacientes dializados y los no dializados. Datos preclínicos sobre seguridad: Carcinogenicidad: La carcinogenicidad de MIRCERA no se ha evaluado en estudios con animales de larga duración. MIRCERA no inducía una respuesta proliferativa en líneas de células tumorales no hematológicas cultivadas in vitro. En un estudio de toxicidad en ratas de seis meses de duración, no se observó ninguna respuesta tumorígena o inesperadamente mitógena en tejidos no hematológicos. Además, en un estudio con diversos tejidos humanos sólo se produjo la unión in vitro de MIRCERA a células diana (células progenitoras de la médula ósea). Trastornos de la fecundidad: Cuando MIRCERA se administró a ratas macho y hembra por vía subcutánea antes del apareamiento y durante el mismo, no afectó a la reproducción, la fertilidad ni los parámetros de evaluación espermática. Teratogenicidad: Los estudios en animales no han revelado ningún efecto nocivo de MIRCERA sobre la preñez, el desarrollo embrional/fetal, el parto ni el desarrollo posnatal.
Indicaciones.
MIRCERA está indicado para el tratamiento de la anemia asociada a insuficiencia renal crónica (IRC) en adultos, incluidos los pacientes dializados y los no dializados.
Dosificación.
Dosis habitual: Gracias a su semivida de eliminación más larga, MIRCERA se administra con menor frecuencia que otros estimuladores de la eritropoyesis (EE). El tratamiento con MIRCERA requiere la supervisión de un profesional sanitario. Tratamiento de pacientes anémicos con insuficiencia renal crónica: La solución puede administrarse por vía subcutánea (s.c.) o intravenosa (i.v.), según la preferencia clínica. MIRCERA puede inyectarse subcutáneamente en el abdomen, un brazo o un muslo. Los tres sitios son igual de adecuados para la inyección subcutánea de MIRCERA. Se recomienda determinar la hemoglobina cada dos semanas hasta su estabilización y periódicamente después. Pacientes no tratados actualmente con un estimulador de la eritropoyesis: Pacientes no dializados: Para elevar la cifra de hemoglobina por encima de 10 g/dl (6,21 mmol/l), la dosis inicial recomendada es de 1,2 mg/kg de peso una vez al mes, en inyección subcutánea. Como alternativa se puede administrar una dosis inicial de 0,6 mg/kg de peso una vez cada dos semanas, en inyección i.v. o s.c. única. Pacientes dializados. Para elevar la cifra de hemoglobina por encima de 10 g/dl (6,21 mmol/l), la dosis inicial recomendada de 0,6 mg/kg de peso puede administrarse una vez cada dos semanas, en inyección i.v. o s.c. única. La dosis de MIRCERA puede incrementarse aproximadamente en un 25% de la dosis anterior si el aumento de la hemoglobina es inferior a 1,0 g/dl (0,621 mmol/l) en el plazo de un mes. También pueden realizarse incrementos ulteriores del mismo orden (25%) a intervalos mensuales hasta alcanzar la concentración deseada de hemoglobina. Si el aumento de la concentración de hemoglobina en un mes es superior a 2 g/dl (1,24 mmol/l), debe reducirse la dosis aproximadamente en un 25%. Si la cifra de hemoglobina supera los 13 g/dl (7,45 mmol/l), debe interrumpirse el tratamiento hasta que descienda a menos de 12 g/dl, para proseguirlo después con una dosis aproximadamente a la mitad de la anterior. En las regiones en las que se ha establecido un límite superior de la hemoglobina de 12 g/dL, debe considerarse reducir o ajustar la dosis en un 25%. Una vez interrumpido el tratamiento, cabe esperar un descenso de la cifra de hemoglobina de aproximadamente 0,35 g/dl por semana. Los pacientes tratados una vez cada dos semanas cuya concentración de hemoglobina sea superior a 10 g/dl (6,21 mmol/l) pueden recibir MIRCERA una vez al mes en una dosis que duplique la anterior administrada una vez cada dos semanas. La dosis no debe ajustarse más de una vez al mes. Pacientes tratados actualmente con un estimulador de la eritropoyesis: Los pacientes tratados actualmente con un EE pueden cambiar a MIRCERA en una dosis mensual en inyección única, administrado en inyección i.v. o s.c. única. La dosis inicial de MIRCERA se establece a partir de la dosis semanal anterior de darbepoetina alfa o epoetina en el momento de la sustitución, según se muestra en las Tablas 1 y 2. La primera inyección de MIRCERA debe administrarse en el momento de la siguiente dosis prevista de darbepoetina alfa o epoetina.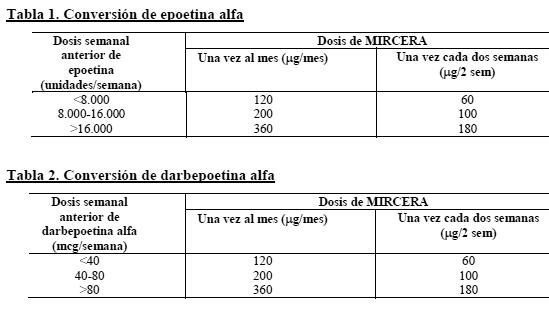
Si se requiere un ajuste posológico para mantener la concentración deseada de hemoglobina por encima de 10 g/dl (6,21 mmol/l), la dosis mensual puede ajustarse en aproximadamente un 25%. Si el aumento de la concentración de hemoglobina en un mes es superior a 2 g/dl (1,24 mmol/l), y ha alcanzado 12g/dl (7,45 mmol/L) debe reducirse la dosis aproximadamente en un 25%. Si la cifra de hemoglobina supera los 13 g/dl (8,07 mmol/l), debe interrumpirse el tratamiento hasta que descienda a menos de 12 g/dl, para proseguirlo después con una dosis aproximadamente un 25% inferior a la dosis administrada previamente. Una vez interrumpido el tratamiento, cabe esperar un descenso de la cifra de hemoglobina de aproximadamente 0,35 g/dl por semana. La dosis no debe ajustarse más de una vez al mes. Interrupción del tratamiento: Por lo general, el tratamiento con MIRCERA es de larga duración. Ahora bien, se puede interrumpir en cualquier momento si es necesario. Dosis no administradas (olvido de dosis): Si no se ha administrado una dosis de MIRCERA, deberá hacerse después lo antes posible. A continuación, se proseguirá la administración de MIRCERA con la frecuencia prescrita. Pautas posológicas especiales: Niños: MIRCERA no está recomendado para pacientes menores de 18 años, pues no hay datos sobre su seguridad y eficacia en este grupo poblacional. Ancianos: no es necesario ajustar la dosis inicial en los pacientes de 65 o más años (ver Ancianos). Insuficiencia hepática: No se ha establecido la seguridad ni la eficacia del tratamiento con Mircera en pacientes con hepatopatía grave. Por lo tanto, se debe tener precaución en estos casos.
Contraindicaciones.
MIRCERA está contraindicado en los siguientes casos: Hipertensión no controlada. Hipersensibilidad al principio activo o a alguno de los excipientes.
Reacciones adversas.
Estudios clínicos: La base de datos de seguridad de MIRCERA correspondiente a los estudios clínicos controlados tenía 3.042 pacientes con IRC, de los que 1.939 recibieron tratamiento con MIRCERA y 1.103 con otros EE. De acuerdo con los resultados obtenidos en estos 1.939 pacientes, cabe esperar reacciones adversas en aproximadamente el 6% de los pacientes tratados con MIRCERA. La reacción adversa más común ha sido la hipertensión (frecuente). Para clasificar la frecuencia de las reacciones adversas atribuidas a MIRCERA en los estudios clínicos controlados se han utilizado los descriptores siguientes: frecuente ( > 1/100 y < 1/10), nada frecuente ( > 1/1.000 y < 1/100) y escasa ( > 1/10.000 y < 1/1.000).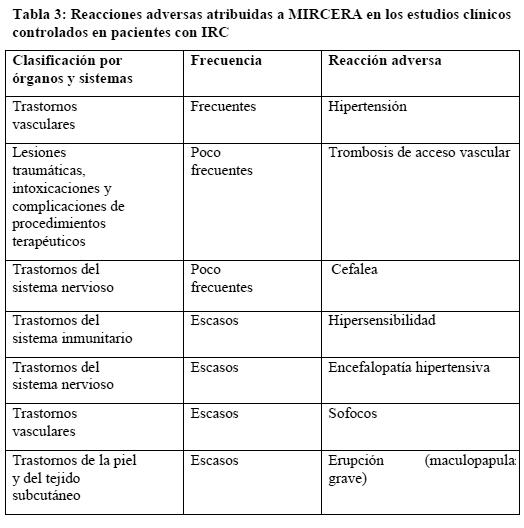
La frecuencia de todos los demás acontecimientos adversos notificados atribuidos a MIRCERA fue escasa y en la mayoría de los casos de intensidad leve o moderada. Estos acontecimientos adversos eran compatibles con la comorbilidad conocida en la población. Alteraciones analíticas: Durante el tratamiento con MIRCERA en los estudios clínicos se observó un leve descenso del recuento plaquetario, pero que permaneció dentro de los valores normales. Se registraron recuentos de plaquetas inferiores a 100 x 109/l en el 5% de los pacientes tratados con MIRCERA y en el 2% de los que habían recibido otros EE. Poscomercialización: Se ha notificado aplasia eritrocitaria pura (AEP) causada por anticuerpos anti-eritropoyetina asociada al tratamiento con MIRCERA Cuando se diagnostique AEP, los pacientes deben interrumpir el tratamiento con MIRCERA y no sustituirlo por otra eritropoyetina recombinante (ver Advertencias). Con esta única excepción, la información sobre seguridad tras la comercialización concuerda con el perfil habitual de acontecimientos adversos en estas poblaciones y el de reacciones adversas de MIRCERA (ver Advertencias, Uso en poblaciones especiales y Estudios clínicos). Alteraciones analíticas: Ver Poscomercialización.
Advertencias.
Se recomiendan suplementos de hierro para todos los pacientes con valores de ferritina sérica por debajo de 100 mg/l o cuya saturación de la transferrina sea inferior al 20%. Para tener la seguridad de una eritropoyesis eficaz, se deben evaluar las reservas de hierro en todos los pacientes antes del tratamiento y durante el mismo. Falta de efecto: Las causas más frecuentes de una respuesta incompleta a los EE son un déficit de hierro y las enfermedades inflamatorias. Los trastornos siguientes también pueden menoscabar la eficacia terapéutica de los EE: infecciones intercurrentes, intoxicación grave por aluminio, pérdida de hueso crónica, fibrosis medular, sobrecarga alumínica grave debida al tratamiento de una insuficiencia renal, déficit de ácido fólico o vitamina B12 y hemólisis. Si pueden excluirse todos los trastornos antedichos y el paciente sufre una caída repentina de la hemoglobina asociada a reticulocitopenia y anticuerpos contra la eritropoyetina, debe considerarse la conveniencia de un examen de la médula ósea para determinar si existe aplasia eritrocitaria pura (AEP). Si se diagnostica una AEP, ha de retirarse MIRCERA y no se debe cambiar a los pacientes a otro EE. AEP: Se ha notificado AEP causada por anticuerpos anti-eritropoyetina en asociación con EE, incluido MIRCERA. Se ha observado que estos anticuerpos tienen una reacción cruzada con todos los EE, por lo que los pacientes con sospecha o confirmación de presentar anticuerpos contra la eritropoyetina no deben cambiarse a MIRCERA. Concentración de hemoglobina: En pacientes con IRC, la concentración de hemoglobina de mantenimiento no debe rebasar el límite superior de la concentración deseada de hemoglobina recomendada en Dosificación. En estudios clínicos se ha observado un aumento del riesgo de fallecimiento y acontecimientos cardiovasculares graves con la administración de agentes estimuladores de la eritropoyesis para alcanzar una concentración de hemoglobina superior a 12 g/dl (7,45 mmol/l). En estudios clínicos controlados no se han demostrado beneficios significativos atribuibles a la administración de epoetinas cuando se ha incrementado la concentración de hemoglobina por encima del valor necesario para controlar los síntomas de la anemia y evitar una transfusión sanguínea. Vigilancia de la tensión arterial: Como con otros EE, la tensión arterial puede elevarse durante el tratamiento de la anemia con MIRCERA. La tensión arterial debe controlarse adecuadamente antes del tratamiento con MIRCERA, al comenzarlo y durante su desarrollo. Si resulta difícil controlar la tensión arterial farmacológicamente o con medidas dietéticas, se reducirá la dosis de MIRCERA o se suspenderá su administración (v. Dosificación). Efecto sobre el crecimiento tumoral: MIRCERA, al igual que otros EE, es un factor del crecimiento que fundamentalmente estimula la producción eritrocitaria. Receptores de la eritropoyetina pueden expresarse en la superficie de diversos tipos de células tumorales. Como con todos los factores de crecimiento, existe la preocupación de que los EE puedan estimular el crecimiento de cualquier tipo de tumor canceroso. En estudios clínicos controlados en los que se administraron epoetinas a pacientes con distintos tipos de cáncer, incluidos los de cabeza y cuello y de mama, hubo una mortalidad excesiva no explicada. MIRCERA no está aprobado para el tratamiento de la anemia en pacientes con cáncer. La seguridad y la eficacia de MIRCERA no se conoce en pacientes con hemoglobinopatías, hepatopatías graves, crisis epilépticas, convulsiones, hemorragias con necesidad de transfusiones o una cifra de plaquetas superior a 500 x 109/l. Por consiguiente, ha de procederse con especial precaución en estos pacientes. El uso indebido de MIRCERA por personas sanas puede llevar a un aumento excesivo de la concentración de hemoglobina, el cual a su vez puede asociarse con complicaciones cardiovasculares potencialmente mortales. Efectos sobre la capacidad para conducir y utilizar máquinas: No se han llevado a cabo estudios sobre los efectos de MIRCERA en la capacidad de conducir vehículos y utilizar maquinaria. Ahora bien, no se espera ningún efecto considerando el mecanismo de acción y el conocido perfil de seguridad toxicológica de MIRCERA.
Interacciones.
No se ha realizado ningún estudio de interacción. Los resultados clínicos no muestran ninguna interacción de MIRCERA con otros medicamentos. El efecto de otros fármacos en la farmacocinética y la farmacodinámica de MIRCERA se ha estudiado mediante un análisis poblacional. Los datos obtenidos no revelan ningún efecto de medicaciones concomitantes en la farmacocinética y la farmacodinámica de MIRCERA. Uso en poblaciones especiales: Embarazo: No hay datos adecuados sobre la utilización de MIRCERA en mujeres embarazadas. Los estudios en animales no han revelado ningún efecto nocivo, directo o indirecto, sobre la preñez, el desarrollo embrio-fetal, el parto o el desarrollo posnatal. No obstante, se procederá con precaución cuando se administre MIRCERA a mujeres embarazadas. Lactancia: No se sabe si la metoxipolietilenglicol-epoetina beta pasa a la leche materna humana. En un estudio con animales se ha observado la excreción de metoxipolietilenglicol-epoetina beta en la leche materna. La decisión entre continuar o suspender la lactancia materna y continuar o suspender el tratamiento con MIRCERA debe tomarse considerando la importancia de la lactancia materna para el niño y el beneficio terapéutico de MIRCERA para la madre. Uso en pediatría: Ver Pautas posológicas especiales. Ancianos: De los 1.789 pacientes con IRC tratados con MIRCERA en estudios clínicos de fase II y III de MIRCERA, el 24% tenían una edad de 65 a 74 años, y el 20%, de 75 o más años. De acuerdo con los análisis poblacionales, no es necesario ajustar la dosis inicial en los pacientes de 65 o más años. Ver Pautas posológicas especiales. Insuficiencia hepática: Ver Pautas posológicas especiales y Farmacocinética en poblaciones especiales.
Conservación.
Este medicamento no debe utilizarse después de la fecha de caducidad, indicada con EXP en el envase. MIRCERA debe conservarse en un refrigerador, a 2-8°C. Manténgase la jeringa precargada en el embalaje exterior para protegerla de la luz. MIRCERA no debe congelarse. El paciente puede conservar MIRCERA fuera del refrigerador, a temperatura ambiente (no más de 30°C), durante un período único de 1 mes. Una vez sacado del refrigerador, MIRCERA ha de utilizarse dentro de este período. Instrucciones especiales de uso, manipulación y eliminación: MIRCERA no debe mezclarse con otros productos. MIRCERA es un producto estéril, pero no contiene conservantes. MIRCERA no debe administrarse en más de una dosis por jeringa precargada o vial. Sólo se inyectará la solución si está límpida, es incolora o ligeramente amarillenta y no contiene ninguna partícula visible. MIRCERA no debe agitarse. El producto debe haber alcanzado la temperatura ambiente antes de inyectarse. Eliminación de medicamentos no utilizados/caducados: La emisión de productos farmacéuticos al medio ambiente debe reducirse a un mínimo. Los medicamentos no deben eliminarse a través de las aguas residuales, y su eliminación con los residuos domésticos también debe evitarse. Utilice los sistemas de recogida establecidos si los hay en su localidad.
Sobredosificación.
El margen de seguridad terapéutica de MIRCERA es amplio. En el tratamiento con MIRCERA ha de considerarse la respuesta individual de cada paciente. Una sobredosis puede manifestarse en un efecto farmacodinámico exagerado, por ejemplo en una eritropoyesis excesiva. Si las cifras de hemoglobina son excesivas, MIRCERA debe retirarse temporalmente (ver Dosificación). Puede realizarse una flebotomía si está clínicamente indicada.
Presentación.
Jeringas precargadas con 50 mg en 0,3 ml: envase con 1. Jeringas precargadas con 75 mg en 0,3 ml: envase con 1. Jeringas precargadas con 100 mg en 0,3 ml: envase con 1. Jeringas precargadas con 150 mg en 0,3 ml: envase con 1. Jeringas precargadas con 200 mg en 0,3 ml: envase con 1. Jeringas precargadas con 250 mg en 0,3 ml: envase con 1.