PERGOVERIS 150 UI/75 UI
MERCK
Pergoveris 150 UI/75 UI polvo y disolvente para solución inyectable.
Composición.
Un vial contiene 150 UI de folitropina alfa (r-hFSH) (equivalente a 11 microgramos) y 75 UI de lutropina alfa (r-hLH) (equivalente a 3,0 microgramos). La solución reconstituida contiene 150 UI de r-hFSH y 75 UI de r-hLH por mililitro. La folitropina alfa y la lutropina alfa se producen en células de ovario de hámster chino (CHO), modificadas por ingeniería genética. Excipientes: sacarosa, fosfato disódico dihidrato, fosfato monosódico monohidrato. Forma farmacéutica: Polvo y disolvente para solución inyectable. Polvo: pastilla liofilizada blanca. Disolvente: solución clara, incolora. El pH de la solución reconstituida es 6,5 - 7,5.
Farmacología.
Propiedades farmacodinámicas: Grupo Farmacoterapéutico: grupo gonadotrofinas, código ATC: G03GA05/G03GA07. Pergoveris es un preparado de hormona foliculoestimulante y hormona luteinizante producidas en células de ovario de hámster chino (CHO), modificadas por ingeniería genética. En los ensayos clínicos la eficacia de la combinación de folitropina alfa y lutropina alfa se ha demostrado en mujeres con hipogonadismo hipogonadotropo. Durante la estimulación del desarrollo folicular en mujeres con anovulación y déficit de LH y FSH, el principal efecto debido a la administración de lutropina alfa es un aumento de la secreción de estradiol por los folículos, cuyo desarrollo es estimulado por la FSH. En un ensayo clínico en pacientes con hipogonadismo hipogonadotropo y un nivel sérico de LH endógena menor de 1,2 UI/l se investigó la dosis adecuada de r-hLH (lutropina alfa). Una dosis de 75 UI de r-hLH administrada diariamente (en combinación con 150 UI de folitropina alfa (r-hFSH)) resultó adecuada para el desarrollo folicular y la producción de estrógenos. Una dosis de 25 UI de rhLH administrada diariamente (en combinación con 150 UI de folitropina alfa) originó un desarrollo folicular insuficiente. Por tanto, la administración de menos de un vial de Pergoveris diariamente puede proporcionar una actividad de LH demasiado baja para asegurar un adecuado desarrollo folicular. Propiedades farmacocinéticas: La combinación de folitropina alfa y lutropina alfa ha demostrado tener el mismo perfil farmacocinético que el de la folitropina alfa y lutropina alfa por separado. Folitropina alfa: Tras la administración intravenosa, la folitropina alfa se distribuye en el espacio extracelular con una semivida de distribución de unas 2 horas y con una semivida de eliminación, de alrededor 1 día. En equilibrio estacionario, el volumen de distribución es de 10 l y el aclaramiento total, de 0,6 l/h, respectivamente. La octava parte de la dosis de folitropina alfa se excreta en la orina. Tras la administración subcutánea, la biodisponibilidad absoluta es de alrededor del 70%. Tras la administración de dosis repetidas de folitropina alfa, se produce una acumulación de 3 veces, alcanzando un equilibrio estacionario en un periodo de 3-4 días. En mujeres con supresión de la secreción endógena de gonadotrofinas, la folitropina alfa estimula adecuadamente el desarrollo folicular y la esteroidogénesis, a pesar de unos niveles indetectables de LH. Lutropina alfa: Tras la administración intravenosa, la lutropina alfa se distribuye rápidamente, con una semivida de distribución de aproximadamente una hora y se elimina del organismo con una semivida de eliminación de unas 10-12 horas. El volumen de distribución en equilibrio estacionario es de alrededor de 10-14 l. La farmacocinética de la lutropina alfa es lineal, ya que el AUC es directamente proporcional a la dosis administrada. El aclaramiento total es de alrededor de 2 l/h, y menos del 5 % de la dosis se excreta en la orina. El tiempo medio de permanencia es de aproximadamente 5 horas. Tras la administración subcutánea, la biodisponibilidad absoluta es aproximadamente del 60%; la semivida de eliminación es algo más prolongada. Tras la administración única o repetida de lutropina alfa, la farmacocinética de la lutropina alfa es comparable y su tasa de acumulación es mínima. No existe interacción farmacocinética con la folitropina alfa cuando se administran simultáneamente. Datos preclínicos sobre seguridad: Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad de dosis repetidas y genotoxicidad.
Indicaciones.
Pergoveris está indicado en mujeres con déficit severo de LH y FSH para la estimulación del desarrollo folicular. En los ensayos clínicos, estas pacientes se eligieron por un nivel sérico de LH endógena de < 1,2 UI/l.
Dosificación.
El tratamiento con Pergoveris debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de problemas de fertilidad. Pergoveris se administra vía subcutánea. El polvo debe reconstituirse, inmediatamente antes de su uso, con el disolvente suministrado. En mujeres con déficit de LH y FSH (hipogonadismo hipogonadotropo), el objetivo del tratamiento con Pergoveris es desarrollar un único folículo de Graaf maduro, a partir del cual se libera el ovocito tras la administración de gonadotropina coriónica humana (hCG). Pergoveris debe administrarse como un ciclo de inyecciones diarias. Puesto que estas pacientes son amenorreicas y tienen una escasa secreción endógena de estrógenos, el tratamiento puede comenzar en cualquier momento. El tratamiento debe adaptarse a la respuesta individual de la paciente, evaluada mediante el tamaño folicular determinado por ecografía y respuesta estrogénica. Una pauta recomendada comienza con un vial de Pergoveris al día. Si se administra menos de un vial de Pergoveris al día, la respuesta folicular puede que no sea satisfactoria porque la cantidad de lutropina alfa sea insuficiente. Si se considera apropiado aumentar la dosis de FSH, el ajuste de dosis debe realizarse preferentemente a intervalos de 7-14 días, con incrementos de 37,5-75 UI y utilizando una folitropina alfa autorizada. Puede aceptarse la prolongación del tiempo de estimulación en un ciclo determinado, hasta 5 semanas. Cuando se obtiene una respuesta óptima, debe administrarse una inyección única de 5.000 UI a 10.000 UI de hCG, 24-48 horas después de la última inyección de Pergoveris. Se recomienda a la paciente que realice el coito el mismo día de la administración de hCG, así como al día siguiente. De forma alternativa, se puede realizar inseminación intrauterina (IIU). Puede considerarse la necesidad de apoyo de fase lútea, ya que la falta de sustancias con actividad luteotropa (LH/hCG) después de la ovulación puede dar lugar a un fracaso prematuro del cuerpo lúteo. Si se obtiene una respuesta excesiva, debe interrumpirse el tratamiento y no administrarse hCG. El tratamiento debe reiniciarse en el ciclo siguiente con una dosis de FSH más baja que la del ciclo previo. En los ensayos clínicos, las pacientes con déficit severo de FSH y LH se definieron por la presentación de niveles séricos de LH endógena < 1,2 UI/l determinado en un laboratorio central. Sin embargo, debe tenerse en cuenta que hay variaciones en las determinaciones de LH realizadas en diferentes laboratorios. En estos ensayos clínicos la tasa de ovulación por ciclo fue de 70-75%.
Contraindicaciones.
Pergoveris está contraindicado en pacientes que presentan: hipersensibilidad a los principios activos, folitropina alfa y lutropina alfa, o a alguno de los excipientes; tumores del hipotálamo o de la hipófisis; aumento del tamaño de los ovarios o quistes no debidos a poliquistosis ovárica; hemorragias ginecológicas de origen desconocido. Carcinoma ovárico, uterino o mamario. Pergoveris no debe utilizarse cuando no pueda obtenerse una respuesta eficaz, en casos tales como: fallo ovárico primario; malformaciones de los órganos sexuales incompatibles con el embarazo; mioma uterino incompatible con el embarazo.
Reacciones adversas.
Las reacciones adversas se presentan en orden decreciente de gravedad dentro de cada intervalo de frecuencia.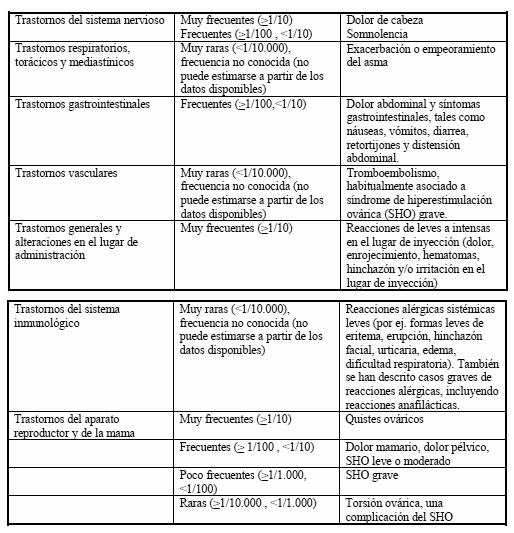
Advertencias.
Pergoveris contiene gonadotropinas potentes capaces de causar reacciones adversas leves o graves, y sólo debe utilizarse por médicos que estén muy familiarizados con los problemas de infertilidad y su tratamiento. El tratamiento con gonadotropinas requiere cierta dedicación por parte de los médicos y profesionales sanitarios, además de disponer de instalaciones de monitorización apropiadas. Para un uso seguro y eficaz de Pergoveris en mujeres, se requiere monitorizar la respuesta ovárica mediante ecografías solas o preferentemente combinadas con la determinación de los niveles séricos de estradiol, de manera regular. Puede existir cierto grado de variabilidad en la respuesta a la administración de FSH/LH entre las pacientes, presentando algunas escasa respuesta a la FSH/LH. En mujeres, se debe utilizar la mínima dosis efectiva para lograr el objetivo del tratamiento. La autoadministración de Pergoveris sólo debe realizarse por pacientes adecuadamente motivadas y entrenadas para ello, con acceso al consejo de un profesional. La primera inyección de Pergoveris debe administrarse bajo supervisión médica directa. Las pacientes con porfiria o con historia familiar de porfiria deben controlarse estrechamente durante el tratamiento con Pergoveris. El deterioro de dicha enfermedad o su aparición por primera vez puede requerir la interrupción del tratamiento. Pergoveris contiene menos de 1 mmol de sodio (23 mg) por dosis, es decir, está prácticamente "libre de sodio". Pergoveris contiene 30 mg de sacarosa por dosis. Esto debe tenerse en cuenta en pacientes con diabetes melitus. Antes de iniciar el tratamiento, debe valorarse adecuadamente el tipo de infertilidad de la pareja y la posible existencia de contraindicaciones en el embarazo. En particular, debe evaluarse lo siguiente: presencia de hipotiroidismo; insuficiencia suprarrenal; hiperprolactinemia y tumores hipofisarios o hipotalámicos. Debe instaurarse el tratamiento específico apropiado. Las pacientes sometidas a estimulación del crecimiento folicular tienen un mayor riesgo de presentar hiperestimulación, debido a la posibilidad de una respuesta estrogénica excesiva y al desarrollo de múltiples folículos. En los ensayos clínicos, la lutropina alfa en combinación con la folitropina alfa ha demostrado que aumenta la sensibilidad ovárica a las gonadotropinas. Si se considera apropiado aumentar la dosis de FSH, el ajuste de dosis debe realizarse preferentemente a intervalos de 7-14 días y con incrementos de 37,5-75 UI utilizando una folitropina alfa autorizada. El síndrome de hiperestimulación ovárica (SHO) es un cuadro clínico distinto al del aumento del tamaño ovárico no complicado. El SHO es un síndrome que puede manifestarse con grados crecientes de gravedad. Incluye un aumento ovárico marcado, niveles elevados de esteroides sexuales y un aumento de la permeabilidad vascular que puede dar lugar a un acúmulo de líquidos en la cavidad peritoneal, pleural y, raramente, pericárdica. En los casos de SHO grave puede observarse la siguiente sintomatología: dolor abdominal; distensión abdominal; aumento importante de los ovarios; aumento de peso; disnea; oliguria y síntomas gastrointestinales incluyendo naúseas, vómitos y diarrea. La evaluación clínica puede revelar: hipovolemia; hemoconcentración; alteraciones del equilibrio electrolítico; ascitis; hemoperitoneo; derrames pleurales; hidrotórax; distrés respiratorio agudo y fenómenos tromboembólicos. Muy raramente, el SHO grave puede complicarse con embolia pulmonar, accidente cerebrovascular isquémico o infarto de miocardio. La respuesta ovárica excesiva raramente da lugar a una hiperestimulación significativa, a no ser que se administre hCG para inducir la ovulación. Por tanto, en caso de hiperestimulación ovárica es prudente no administrar hCG, advirtiendo a la paciente que no realice el coito o que utilice métodos anticonceptivos de barrera durante al menos 4 días. El SHO puede progresar rápidamente (en menos de 24 horas o en varios días) hasta convertirse en un cuadro clínico grave, por lo que debe seguirse a las pacientes durante al menos dos semanas tras la administración de hCG. Para minimizar el riesgo de SHO o de embarazo múltiple (ver a continuación), se recomienda practicar ecografías, así como determinaciones de estradiol. En caso de anovulación, el riesgo de SHO aumenta si existe un nivel sérico de estradiol > 900 pg/ml (3300 pmol/l) y si hay más de 3 folículos con un diámetro igual o superior a 14 mm. La incidencia del síndrome de hiperestimulación ovárica y embarazos múltiples puede minimizarse utilizando la dosis y el esquema posológico de Pergoveris y FSH recomendados y monitorizando cuidadosamente el tratamiento (ver a continuación). El SHO puede ser más grave y más prolongado si se produce embarazo. Muy a menudo el SHO se produce después de interrumpir el tratamiento hormonal y alcanza su máxima intensidad al cabo de siete a diez días después del tratamiento. Habitualmente, el SHO se resuelve espontáneamente al comenzar la menstruación. Si se produce SHO grave, debe interrumpirse el tratamiento con gonadotropinas si es que todavía continúa. Debe hospitalizarse a la paciente e iniciar el tratamiento específico del SHO. La incidencia de este síndrome es mayor en pacientes con poliquistosis ovárica. En pacientes sometidas a inducción de la ovulación, la incidencia de embarazos múltiples es más elevada que en los casos de concepción natural. La mayoría de embarazos múltiples son gemelares. Para minimizar el riesgo de embarazo múltiple, se recomienda una monitorización cuidadosa de la respuesta ovárica. Antes de iniciar el tratamiento se debe informar a las pacientes del riesgo potencial de tener embarazos múltiples. La incidencia de fracaso del embarazo debido a aborto en pacientes sometidas a estimulación del desarrollo folicular para inducir la ovulación o practicar ART es superior a la observada en la población general. Cuando se presente riesgo de SHO o de embarazos múltiples, debe considerarse la posibilidad de interrupir el tratamiento. Las mujeres con historia de enfermedad tubárica presentan riesgo de embarazo ectópico, tanto si el embarazo es por concepción espontánea como si se logra mediante tratamientos de fertilidad. Se ha descrito que la prevalencia del embarazo ectópico tras practicar FIV es del 2%-5%, en comparación con un 1%-1,5% en la población general. Se han notificado neoplasias de ovario y de otros órganos del aparato reproductor, tanto benignas como malignas, en mujeres sometidas a múltiples tratamientos de infertilidad. Todavía no está establecido si el tratamiento con gonadotropinas aumenta o no el riesgo habitual de estos tumores en mujeres infértiles. La prevalencia de malformaciones congénitas tras ART puede ser ligeramente superior a la observada tras la concepción natural. Esto se considera debido a diferencias en las características de los progenitores (por ejemplo, la edad de la madre o las características del semen) y a los embarazos múltiples. En mujeres con factores de riesgo generalmente reconocidos de presentar problemas tromboembólicos, tales como historia familiar o personal, el tratamiento con gonadotropinas puede aumentar dicho riesgo. En estas mujeres, los beneficios de la administración de gonadotropinas deben sopesarse frente a los riesgos. No obstante, hay que tener en cuenta que el embarazo por sí mismo también comporta un aumento del riesgo de fenómenos tromboembólicos.
Interacciones.
Pergoveris no debe mezclarse con otros medicamentos en la misma jeringa, excepto con folitropina alfa. Embarazo y lactancia: Pergoveris no debe administrarse durante el embarazo o la lactancia. Efectos sobre la capacidad para conducir y utilizar máquinas: No se han realizado estudios sobre la capacidad para conducir y utilizar máquinas.
Conservación.
No conservar a temperatura superior a 25°C. Conservar en el embalaje exterior para protegerlo de la luz. Incompatibilidades: Este medicamento no debe mezclarse con otros excepto con los mencionados en la sección. Precauciones especiales de eliminación: Período de validez: 3 años. Para uso único e inmediato tras la primera apertura y reconstitución. Precauciones especiales de eliminación: Para uso único. Pergoveris debe reconstituirse con el disolvente antes de su utilización. La solución reconstituida no debe administrarse si contiene partículas o no es nítida. Pergoveris puede mezclarse con folitropina alfa y coadministrarse en una misma inyección. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con las normativas locales. Presentación: Cada envase contiene 2 viales para reconstituir.
Sobredosificación.
Los efectos de una sobredosis de Pergoveris son desconocidos. Sin embargo, puede esperarse que se produzca un síndrome de hiperestimulación ovárica, que se describe más ampliamente en la sección Advertencias y precauciones especiales de empleo.
Presentación.
Polvo: viales de 3 ml (de vidrio tipo I) que se cierran con un tapón de bromobutilo protegidos por anillos de aluminio y cápsulas flip-off. 1 vial contiene 11 microgramos de r-hFSH y 3 microgramos de r-hLH. Disolvente: viales de 3 ml (de vidrio tipo I) con un tapón de goma recubiertos de Teflon protegido por anillos de aluminio y cápsulas flip-off. 1 vial de disolvente contiene 1 ml de agua para inyección. El medicamento se suministra en cajas que contienen 1, 3 ó 10 viales, con el correspondiente número de viales de disolvente. Puede que solamente estén comercializados algunos tamaños de los envases.