PROLIFT®
PFIZER
Antidepresivo.
Composición.
Comprimidos contienen 4 mg de reboxetina (como metansulfonato). Excipientes: Fosfato de calcio dibásico dihidrato, Celulosa microcristalina, Crospovidona, Estearato de magnesio, Dióxido de silicio, c.s.
Toxicología.
Reboxetina no indujo mutaciones genéticas en células bacterianas o de mamíferos in vitro, pero indujo aberraciones cromosómicas en linfocitos humanos in vitro. Reboxetina no causó daño al ADN en células de levadura o de hepatocitos de rata in vitro. En un ensayo in vivo de micronúcleos de ratón, reboxetina no causó daño cromosómico, y no aumentó la incidencia de tumores en los estudios de carcinogénesis en ratas y ratones. Fue reportado hemosiderosis en estudios de toxicidad en ratas solamente. Los estudios en animales no han demostrado ningún efecto teratogénico o cualquier otro efecto sobre la función reproductora global. Las dosis que produjeron concentraciones plasmáticas dentro del rango terapéutico para los seres humanos inducen una alteración del crecimiento y desarrollo y cambios a largo plazo del comportamiento de las crías de ratas. En ratas, reboxetina es excretada en la leche.
Indicaciones.
La Reboxetina está indicada para el tratamiento de la enfermedad depresiva y para mantener la mejoría clínica en los pacientes que responden inicialmente al tratamiento. La remisión de la fase aguda de la enfermedad depresiva está asociada con una mejoría en la calidad de vida del paciente en términos de adaptación social.
Dosificación.
Prolift es para administración oral. El inicio del efecto clínico se ve generalmente después de 14 días de iniciado el tratamiento. Uso en adultos: La dosis terapéutica recomendada es de 4 mg dos veces al día (8 mg/día), administrada oralmente. Esta dosis puede incrementarse hasta 12 mg al día en caso de una respuesta clínica incompleta después de 3 semanas. Uso en ancianos (edad mayor a 65 años): La dosis terapéutica recomendada es de 2 a 3 mg dos veces al día (4 a 6 mg/día) administradas por vía oral. Esta dosis puede aumentarse hasta 6 mg/día en caso que la respuesta clínica sea incompleta después de 3 semanas de iniciado el tratamiento con reboxetina. Uso en niños: No se dispone de datos sobre el uso de reboxetina en niños (ver Advertencias). Uso en pacientes con insuficiencia renal o hepática: La dosis de inicio en pacientes con insuficiencia renal o hepática de moderada a severa, debe ser de 2 mg dos veces al día, la cual puede incrementarse sobre la base de la tolerancia del paciente.
Contraindicaciones.
Hipersensibilidad a reboxetina o cualquier otro componente del producto.
Reacciones adversas.
En estudios controlados con placebo de 8 semanas de duración o menos, se reportaron efectos adversos en aproximadamente 80% de los pacientes tratados con reboxetina y en aproximadamente 70% de los pacientes tratados con placebo. El índice de suspensión para los eventos adversos fueron aproximadamente el 9% y el 5% entre los pacientes tratados con reboxetina y los pacientes tratados con placebo respectivamente.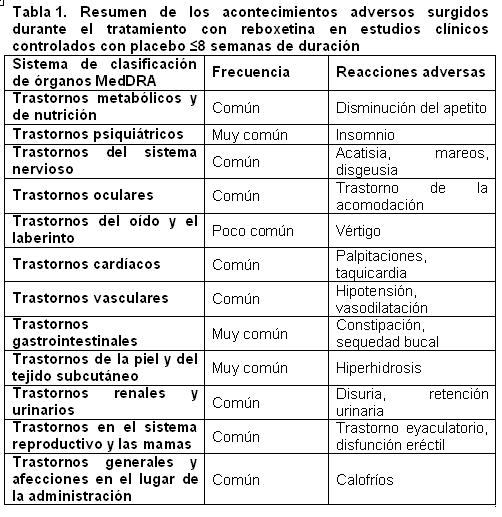
La única modificación en los signos vitales fue un incremento en la frecuencia cardiaca en posición erguida. Aparte de la taquicardia no se observaron cambios consistentes en los trazos del ECG durante el tratamiento con reboxetina en pacientes adultos. En estudios de más de 8 semanas, se reportaron nuevos eventos adversos que surgieron en aproximadamente 30% de los pacientes tratados con reboxetina y en aproximadamente 25% de los pacientes tratados con placebo. El perfil de eventos adversos para los estudios de más de 8 semanas fue coherente con los estudios de hasta 8 semanas. Estos eventos adversos estuvieron asociados con los índices de suspensión de 4% y 1%, respectivamente. El estreñimiento fue el único evento que se observó más comúnmente en el grupo tratado con reboxetina. En aproximadamente el 5% de los pacientes tratados con reboxetina y aproximadamente el 4% de los tratados con placebo ocurrieron acontecimientos adversos después de la interrupción del tratamiento. Vigilancia post-comercialización: En la tabla 2, se muestran los eventos adversos que se han reportado posterior a la comercialización con reboxetina: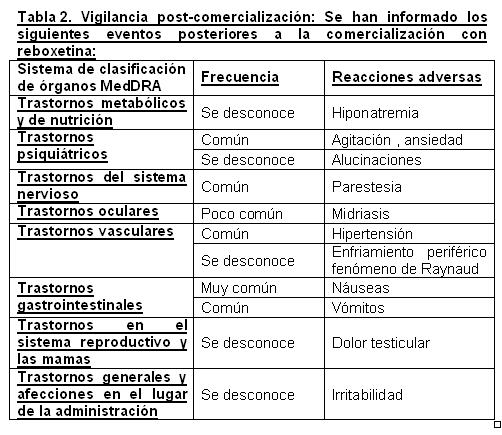
Advertencias.
Debido a que se han reportado raros casos de ataques convulsivos en los estudios clínicos, reboxetina debe administrarse bajo una estrecha supervisión a los sujetos con antecedentes de enfermedades convulsivas y debe suspenderse si el paciente desarrolla ataques. Se debe evitar el uso combinado de los inhibidores de la MAO y reboxetina (ver Interacciones). Igual que con todos los antidepresivos, se han presentado cambios a estados de manía/hipomanía. Se recomienda, por lo tanto, una estrecha supervisión de los pacientes bipolares. En un análisis de 24 estudios clínicos a corto plazo (4 meses), que involucraron a 4400 pacientes niños con depresión mayor, desorden obsesivo-compulsivo u otras alteraciones psiquiátricas, placebos controlados, quienes fueron tratados con antidepresivos inhibidores selectivos de la recaptación de serotonina y antidepresivos de otra clase, se observó un incremento del doble en riesgo de suicidio del grupo que recibió el antidepresivo versus el grupo que recibió placebo (4% versus 2%). El riesgo de un intento suicida es inherente en la depresión y puede persistir hasta que ocurre una remisión significativa; por tanto, la supervisión cercana del paciente durante la terapia inicial con el fármaco es recomendable. Antes de iniciar una terapia con un antidepresivo se deben investigar cuidadosamente los antecedentes psiquiátricos del paciente, incluyendo historia familiar y personal de suicidios y desorden bipolar. Este medicamento no debe administrarse a menores de 18 años de edad. Se ha visto que el uso de antidepresivos en niños y adolescentes aumenta el riesgo de pensamientos y conductas suicidas. Debe aplicarse una estrecha supervisión en pacientes con evidencia actual de retención urinaria y glaucoma. En dosis mayores de la máxima recomendada, se ha observado hipotensión ortostática con mayor frecuencia. Debe ponerse una especial atención cuando se administre Reboxetina con otros fármacos conocidos que bajan la presión sanguínea. Se han informado casos de midriasis asociada al consumo de reboxetina. Por esta razón, se debe tener precaución al recetar reboxetina a pacientes con presión intraocular elevada o a aquellos con riesgo de glaucoma agudo de ángulo estrecho. Uso en niños y adolescentes menores de 18 años de edad: Reboxetina no debería utilizarse en el tratamiento de niños y adolescentes menores de18 años de edad. Los comportamientos suicidas (intentos de suicidio y pensamientos suicidas), y hostilidad (predominantemente agresión, comportamiento oposicional e irritación) fueron más frecuentemente observados en los ensayos clínicos con niños y adolescentes tratados con antidepresivos en comparación con aquellos tratados con placebo. Si, según necesidad clínica, no obstante se toma la decisión de tratar, el paciente debe ser cuidadosamente monitoreado por la aparición de síntomas suicidas. Además, se carece de datos de seguridad a largo plazo en niños y adolescentes en relación con el crecimiento, la maduración, el desarrollo cognitivo y del comportamiento. Uso en adultos jóvenes (18-25 años de edad): Un análisis adicional de los datos agrupados de los antidepresivos disponibles en la actualidad mostró un aumento del riesgo de pensamientos y conductas suicidas en comparación con el placebo en adultos jóvenes en los estudios a corto plazo del trastorno depresivo mayor (TDM) y otros trastornos psiquiátricos. En la actualidad, los datos son insuficientes para cuantificar un aumento del riesgo de pensamientos y conductas suicidas asociadas al tratamiento con reboxetina. Sin embargo, cualquiera que esté considerando el uso de reboxetina en adultos jóvenes debe equilibrar este potencial riesgo con la necesidad clínica. Suicidio / pensamientos suicidas o empeoramiento clínico: La depresión se asocia con un mayor riesgo de pensamientos suicidas, autolesiones y suicidio (eventos relacionados con suicidios). Este riesgo persiste hasta que se produzca una remisión significativa. Debido a que la mejoría puede no ocurrir durante las primeras semanas o más de tratamiento, los pacientes deben ser estrechamente monitoreados hasta que se produzca dicha mejoría. Se debería acompañar la terapia, especialmente al comienzo del tratamiento y tras los cambios de dosis, con una supervisión cercana de los pacientes, en particular aquellos con alto riesgo. Los pacientes (y sus cuidadores) deberían ser alertados sobre la necesidad de controlar cualquier empeoramiento clínico, comportamiento o pensamientos suicidas y cambios inusuales en el comportamiento y acudir inmediatamente al médico si se presentan estos síntomas. Embarazo: No hay datos disponibles de ensayos clínicos sobre la exposición a reboxetina durante el embarazo. Sin embargo, los datos de seguridad post-comercialización en un número muy limitado de embarazos no indican efectos adversos de reboxetina en el embarazo ni en la salud del feto o del recién nacido. Los estudios en animales, en general, no indican efectos nocivos directos o indirectos sobre el embarazo, el desarrollo fetal/embrional o el parto. Algunos deterioros del crecimiento y del desarrollo se han observado en ratas recién nacidas. Reboxetina sólo debe usarse en el embarazo si los beneficios potenciales del tratamiento para la madre superan los posibles riesgos para la desarrollo del feto. Lactancia: Se sabe que reboxetina se excreta en la leche materna. Se puede prever que el nivel de la sustancia activa transferida a la leche materna es muy bajo, sin embargo no hay suficiente información para excluir un riesgo para el lactante. El uso de reboxetina durante la lactancia materna puede considerarse si los beneficios potenciales superan los riesgos para el infante.
Interacciones.
Los estudios in vitro han mostrado que reboxetina no inhibe la actividad de las siguientes isoenzimas del citocromo P450: CYP1A2, CYP2C9, CYP2C19 y CYP2E1. En concentraciones altas, reboxetina inhibe la CYP2D6, pero el significado clínico de esta observación es desconocido. Estudios in vitro demuestran que reboxetina es un inhibidor muy débil de la CYP3A4. Los estudios de metabolismo in vitro indican que reboxetina es metabolizada principalmente por la isoenzima CYP3A4 del citocromo P450; reboxetina no es metabolizada por CYP2D6. Por tanto, se podría esperar que los compuestos que disminuyen la actividad de la CYP3A4, aumenten las concentraciones plasmáticas de reboxetina. En un estudio en voluntarios sanos, ketoconazol, un potente inhibidor de CYP3A4, aumentó las concentraciones plasmáticas de los enantiómeros de reboxetina en aproximadamente un 50%. Se han informado niveles séricos bajos de reboxetina al administrarla conjuntamente con inductores de CYP3A4 tales como fenobarbital y carbamazepina. No se ha encontrado ninguna interacción farmacocinética recíproca entre reboxetina y lorazepam. En un estudio in vivo de dosis múltiples, realizado en voluntarios sanos, se observó una interacción no clínicamente significativa entre fluoxetina y reboxetina. En voluntarios sanos, reboxetina no parece potenciar el efecto del alcohol sobre las funciones cognitivas. El uso concomitante reboxetina con otros antidepresivos (tricíclicos, inhibidores de la MAO, incluyendo linezolid [un antibiótico IMAO reversible no selectivo] y azul de metileno SSRI's y Litio) no ha sido evaluado durante los estudios clínicos. El grado de absorción reboxetina no está significativamente influenciado por la ingestión concomitante de alimentos.
Sobredosificación.
En unos pocos casos, se administraron dosis mayores de las recomendadas a pacientes (12 mg a 20 mg/día) durante un período que va de unos pocos días a algunas semanas durante los estudios clínicos. Los eventos adversos que surgieron por el tratamiento incluyen hipotensión postural, ansiedad e hipertensión. Se han reportado dos casos de auto-sobredosis hasta con 52 mg de Reboxetina. Ningún efecto adverso serio fue observado. En caso de sobredosis, se recomienda una estrecha supervisión, incluyendo el monitoreo de la función cardiaca y los signos vitales.
Presentación.
PROLIFT 4 mg x 20 comprimidos.