RAPAMUNE®
WYETH
Inmunosupresor.
Composición.
Rapamune (sirolimus) es un agente inmunosupresor. Sirolimus es una lactona macrocíclica producida por Streptomyces hygroscopicus. El nombre químico de sirolimus (también conocido como rapamicina) es (3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-9,10,12,13,14,21,22,23,24,25,26,27,32,33,34,34a-hexadecahidro-9,27-dihidroxi-3-[(1R)-2-[(1S,3R,4R)-4-hidroxi-3-metoxiciclohexil]-1-metiletil]-10,21-dimetoxi-6,8,12,14,20,26-hexametil-23,27-epoxi-3H-pirido[2,1-c][1,4] oxaazaciclohentriacontina -1,5,11,28,29 (4H,6H,31H)-pentona. Rapamune® está disponible para administración como una solución oral, que contiene 1 mg/ml de sirolimus y grageas que contienen 0,5; 1 y 2 mg de sirolimus. Los ingredientes inactivos de Rapamune® Solución Oral son Phosal 50 PG (ascorbilpalmitato, propilénglicol, monodiglicéridos, etanol, ácidos grasos de soya, Fosfatidilcolina) y Polisorbato 80, NF. Rapamune Solución oral contiene 1,5% - 2,5% de etanol. Los ingredientes inactivos en Rapamune® grageas incluyen Lactosa monohidrato, macrogol 8000 pulverizado, estearato de magnesio, talco. Macrogol 20.000, monooleato de glicerilo, barniz farmacéutico, sulfato de calcio anhidro, celulosa microcristalina, sucrosa, dióxido de titanio. Poloxámero 188, sucrosa, celulosa microcristalina, Povidona K 29/32, dl-a-tocoferol. Sucrosa, Dióxido de titanio, Óxido de hierro amarillo, Óxido de hierro marrón, Povidona K 29/32. Cera carnauba, Tinta de impresión roja Opacode S-1-15095 (Composición tinta de impresión roja Opacode S-1-15095: Óxido de hierro rojo, Goma laca, Alcohol Isopropílico, Propilenglicol, Simeticona emulsión al 30%).
Toxicología.
Carcinogenicidad: Se llevaron a cabo estudios de carcinogenicidad en ratones y ratas. En el estudio en ratones hembra de 86 semanas de duración con 4 dosificaciones aproximadamente 16 a 135 veces las dosis clínicas (ajustadas por área de superficie corporal) se registró un aumento estadísticamente significativo en la incidencia de linfomas malignos con todas las dosis en comparación con los controles. En un segundo estudio en ratones a dosis que fueron aproximadamente 3 a 16 veces la dosis clínica (ajustado al área de superficie corporal) el adenoma hepatocelular y el carcinoma en machos fueron considerados relacionados a sirolimus. El estudio en ratas de 104 semanas de duración con dosis aproximadamente 0,4 a 1 vez las dosis clínicas (ajustadas por área de superficie corporal) reveló un incremento estadísticamente significativo en la incidencia de adenoma testicular en el grupo que recibió la dosis más alta. Mutagenicidad: El sirolimus no demostró ser genotóxico en el ensayo in vitro de mutación inversa bacteriana ensayo de aberraciones cromosómicas en células ováricas de hámster chino, ensayo de mutación de avance en células de linfoma de ratones o en el ensayo in vivo de micronúcleos en ratones. Toxicología reproductiva: El sirolimus resultó embrio y fetotóxico en ratas con dosis de 0,1 mg/kg y superiores (aproximadamente de 0,2 a 0,5 veces las dosis clínicas ajustadas por área de superficie corporal). La toxicidad embrionaria/fetal se manifestó en mortalidad y pesos fetales reducidos (con retardos asociados en la osificación esquelética). Sin embargo, no se evidenció teratogénesis. En combinación con CsA, las ratas registraron mayor mortalidad embriofetal en comparación con sirolimus solo. No se observaron efectos sobre el desarrollo en conejos con dosis maternas tóxicas de 0,05 mg/kg (aproximadamente de 0,3 a 0,8 veces las dosis clínicas ajustadas por área de superficie corporal). No se observó efecto alguno sobre la fertilidad en ratas hembra después de la administración de dosis de hasta 0,5 mg/kg de sirolimus (aproximadamente 1 a 3 veces las dosis clínicas ajustadas por área de superficie corporal). En ratas macho, se produjo una ligera reducción de la fertilidad comparación con los controles con dosis de 2 mg/kg (aproximadamente 4 a 11 veces las dosis clínicas ajustadas por área de superficie corporal). Un segundo estudio falló al confirmar estos hallazgos. Se observaron reducciones en los pesos testiculares y/o lesiones histológicas (por ej. atrofia tubular y células tubulares gigantes) en ratas después de dosis de 0,65 mg/kg (aproximadamente 1 a 3 veces las dosis clínicas ajustadas por área de superficie corporal) y superiores y en un estudio en monos con 0,1 mg/kg (aproximadamente de 0,4 a 1 vez las dosis clínicas ajustadas por área de superficie corporal) y dosis superiores. Los recuentos espermáticos se redujeron en ratas macho después de la administración de sirolimus durante 13 semanas con dosis de 6 mg/kg (aproximadamente 12 a 32 veces las dosis clínicas ajustadas por área de superficie corporal), pero mejoraron 3 meses después de interrumpir la administración.
Indicaciones.
Rapamune® está indicado para la profilaxis de rechazo de órganos en pacientes mayores de13 años receptores de trasplante renal. Se recomienda monitoreo terapéutico para todos los pacientes que reciben Rapamune®. En pacientes con riesgo inmunológico leve a moderado, se recomienda administrar Rapamune® en un régimen con CsA y corticoides como tratamiento inicial. La suspensión de la CsA puede ser considerada 2 a 4 meses después del transplante. En pacientes con alto riesgo inmunológico (definidos como: receptores de raza negra y/o receptores de un nuevo trasplante, que perdieron uno previo por causa inmunológica y/o pacientes con anticuerpos panel-reactivos elevados (PRA; con un nivel de PRA > 80%), se recomienda que Rapamune se use en combinación de tacrolimus y corticosteroides o ciclosporina y corticoides durante el primer año después del trasplante. La seguridad y eficacia de estas combinaciones en pacientes de alto riesgo de trasplante renal no han sido estudiadas más allá de un año. Por lo tanto, después del primer año tras el trasplante, cualquier ajuste en el régimen inmunosupresor deberá considerarse en base al estado clínico del paciente.
Dosificación.
La biodisponibilidad no ha sido determinada para las grageas que han sido trituradas, masticadas o partidas y por lo tanto esto no puede ser recomendado. A los pacientes que no están en condiciones de tomar las grageas se les deberá prescribir la solución e instruirlos en su uso. Se ha demostrado que dos miligramos (2 mg) de Rapamune Solución Oral son clínicamente equivalentes a 2 mg de Rapamune grageas; por ésto, son intercambiables sobre la base de mg a mg. Sin embargo, se desconoce sí altas dosis de Rapamune Solución Oral son clínicamente equivalentes a altas dosis de Rapamune Grageas sobre la base de mg a mg. Rapamune® debe ser recetado sólo por médicos con experiencia en el tratamiento inmunosupresor y manejo de pacientes con trasplante de órganos. Los pacientes tratados con el medicamento deben ser atendidos en servicios dotados de equipamiento y personal con adecuados recursos médicos de apoyo y de laboratorio. El médico responsable del tratamiento de mantención deberá contar con información completa, indispensable para el seguimiento del paciente. Pacientes con riesgo inmunológico leve a moderado: Tratamiento combinado de Rapamune® y ciclosporina (CsA): Rapamune® debe administrarse oralmente una vez al día. La dosis inicial de Rapamune® tratamiento combinado con Rapamune® y CsA debe administrarse después del transplante tan pronto como sea posible. En receptores de transplante de novo, debe darse una dosis de carga de Rapamune® correspondiente a 3 veces la dosis de mantención. Se recomienda una dosis diaria de mantención de 2 mg en pacientes con trasplante renal, con una dosis de carga de 6 mg. Aunque una dosis de mantención de 5 mg, con una dosis de carga de 15 mg, fue empleada en los estudios clínicos y se demostró su seguridad y su eficacia, no pudo establecerse ninguna ventaja en eficacia sobre la dosis de 2 mg en los pacientes con transplante renal. Los pacientes que recibieron 2 mg de Rapamune® Solución Oral por día demostraron un perfil general de seguridad mejor que los pacientes que recibieron 5 mg de Rapamune® Solución Oral por día. Se recomienda administrar inicialmente Rapamune solución oral y grageas en un régimen con CsA y corticoides. La CsA debería retirarse 2 a 4 meses después del trasplante renal en pacientes con riesgo inmunológico bajo a moderado y la dosis de Rapamune debería ser aumentada para alcanzar las concentraciones sanguíneas recomendadas. El retiro de la CsA no ha sido estudiado en pacientes con rechazo agudo o rechazo vascular Banff 93 grado III antes del retiro de la CsA, ni en aquellos que son diálisis dependientes, o con creatinina sérica > 4,5 mg/dL, tampoco en pacientes de raza negra, re-trasplantados, trasplantes multi-organos o pacientes con un alto panel de anticuerpos reactivos (ver Indicaciones y Eficacia clínica). Rapamune® después de la suspensión de CsA (Denominado Régimen de mantención con Rapamune®, RMR): Los pacientes deberán estar recibiendo tratamiento inicial combinado de Rapamune® y CsA. Entre 2 y 4 meses después del trasplante, deberá ir reduciéndose gradualmente la CsA durante un período de 4 a 8 semanas y ajustándose la dosis de Rapamune® para obtener concentraciones mínimas en sangre de 16 a 24 ng/ml (método cromatográfico) durante el primer año después del trasplante. Luego las concentraciones objetivo de sirolimus deberán ser de 12 a 20 ng/ml (método cromatográfico). Las observaciones realizadas al año y a los 5 años (ver más abajo) se aproximaron a estos rangos. El monitoreo de la concentración terapéutica no debe constituir la única base para ajustar el tratamiento con Rapamune®. Deberá prestarse especial atención a los signos/síntomas, biopsias de tejido y parámetros de laboratorio. La CsA inhibe el metabolismo y transporte del sirolimus y, en consecuencia, las concentraciones de sirolimus disminuirán al suspender la CsA si no se aumenta la dosis de Rapamune®. Pacientes con alto riesgo inmunológico: Tratamiento combinado de Rapamune: Se recomienda que Rapamune se utilice en una combinación de tacrolimus y corticosteroides o ciclosporina y corticosteroides durante el primer año posterior al trasplante en pacientes con alto riesgo inmunológico (definidos como receptores de trasplante de raza negra y/o receptores de un trasplante renal repetido que perdieron un aloinjerto previo por motivos inmunológicos y/o pacientes con un alto panel reactivo de anticuerpos [PRA; nivel máximo de PRA > 80%]). La seguridad y eficacia de estas combinaciones en pacientes con trasplante renal de alto riesgo no han sido estudiadas más allá de un año. Por lo tanto, después del primer año del trasplante, se deberá considera cualquier ajuste en el régimen inmunosupresor en base al estado clínico del paciente. Para los pacientes que reciben Rapamune con tacrolimus, el tratamiento con Rapamune deberá iniciarse con una dosis de carga de hasta 10 mg los días 1 y 2 posteriores al trasplante. A partir del día 3, se deberá administrar una dosis inicial de mantención de 5 mg/día. Se deberá obtener una concentración mínima entre los días 5 y 7, a partir de entonces, se deberá ajustar la dosis diaria de Rapamune para lograr concentraciones mínimas de sirolimus en sangre total de 10-15 ng/mL. Para los pacientes que reciben Rapamune con ciclosporina, el tratamiento con Rapamune deberá iniciarse con una dosis de carga de hasta 15 mg el primer día posterior al trasplante. A partir del día 2, se deberá administrar una dosis inicial de mantención de 5 mg/día. Se deberá obtener una concentración mínima entre los días 5 y 7, a partir de entonces, se deberá ajustar la dosis diaria de Rapamune para lograr concentraciones mínimas de sirolimus en sangre total de 10-15 ng/mL. La dosis inicial de tacrolimus deberá ser de hasta 0,2 mg/kg/día administrados en dosis divididas y la dosis se deberá ajustar para alcanzar concentraciones mínimas en sangre totales de 10-15 ng/ml durante 14 días, de 5-10 ng/ml desde el día 15 hasta el final de la semana 26, y de 3-5 ng/ml desde la semana 27 hasta el final de la semana 52. Se deberá administrar Prednisona a una dosis mínima de 5 mg/día. La dosis inicial de ciclosporina deberá ser de hasta 7 mg/kg/día en dosis divididas y, posteriormente, la dosis deberá ajustarse para alcanzar concentraciones mínimas en sangre total de 200-300 ng/ml durante 14 días, 150-200 ng/ml desde el día 15 hasta el final de la semana 26, y 100-150 ng/ml desde la semana 27 hasta el final de la semana 52. Se deberá administrar Prednisona a una dosis un mínima de 5 mg/día. Se puede utilizar una terapia de inducción de anticuerpos. Uso de Rapamune en todos los receptores de aloinjerto renal: La dosis inicial de Rapamune debería ser administrada tan pronto como sea posible después del trasplante. Ajustes frecuentes de la dosis de Rapamune basados en las concentraciones en estado no estacionario de sirolimus pueden provocar una sobredosis o una subdosis porque sirolimus tiene una larga vida media. Una vez que la dosis de mantención está ajustada, los pacientes deben conservar la nueva dosis de mantención al menos por 7 a 14 días antes de un nuevo ajuste de dosis con el monitoreo de la concentración. En la mayoría de los pacientes el ajuste de dosis se puede basar en una proporción simple: Nueva dosis de Rapamune = dosis actual x (concentración requerida / concentración actual). Una dosis de carga debería ser considerada, además de una nueva dosis de mantención cuando es necesario aumentar considerablemente las concentraciones mínimas de sirolimus: Dosis de carga de Rapamune = 3 x (nueva dosis de mantención - dosis de mantención actual). La máxima dosis de Rapamune administrada en un día no debe exceder los 40 mg. Si una dosis diaria estimada excede los 40 mg debidos a la suma de una dosis de carga, la dosis de carga deberá ser administrada en 2 días. La concentración mínima de Sirolimus debe ser monitoreada al menos entre 3 a 4 días después de una dosis de carga. A los efectos de minimizar la variabilidad de la exposición a Rapamune®, este medicamento debe tomarse siempre de la misma manera, ya sea con o alejado de las comidas. El jugo de pomelo reduce el metabolismo del sirolimus mediado por CYP3A4 y puede aumentar el contratransporte mediado por la P-glucoproteína (P-gp) desde los enterocitos del intestino delgado. En consecuencia, no debe consumirse jugo de pomelo con Rapamune® o emplearse para la dilución de la solución oral. Se recomienda tomar Rapamune 4 horas después de la administración de ciclosporina microemulsión [ciclosporina MODIFICADA]. Ajustes a la dosis: La dosificación inicial para pacientes > 13 años de edad que pesen menos de 40 kg debe ajustarse, en base al área de superficie corporal, a 1 mg /m2 /día. La dosis de carga debe ser de 3 mg /m2. Se recomienda que la dosis de mantención de Rapamune® sea reducida en aproximadamente un tercio en pacientes con deterioro hepático. Sin embargo, en estos casos no es necesario modificar la dosis de carga de Rapamune®. La dosificación no necesita ajustarse debido a función renal deteriorada. Modo de Administración: Rapamune® debe administrarse únicamente por vía oral. Rapamune® debe tomarse siempre de la misma manera, ya sea con o alejado de las comidas para minimizar la variación en la absorción de la droga. Rapamune® solución oral debe diluirse únicamente con agua o jugo de naranja, empleando para ello solamente vasos de vidrio o plástico. Rapamune® solución oral no debe diluirse con jugo de pomelo (ver Interacciones) u otros líquidos. Rapamune® solución oral contiene polisorbato-80, conocido por aumentar la velocidad de extracción de di-(2-etilhexil)ftalato (DEHP) del cloruro de polivinilo (PVC). Esto debería ser tenido en cuenta durante la preparación y administración de Rapamune® solución oral. Es importante que las recomendaciones en Posología y Forma de Administración se sigan estrictamente.
Contraindicaciones.
Rapamune® está contraindicado en pacientes con hipersensibilidad a sirolimus o sus derivados o a cualquier componente del producto farmacéutico.
Reacciones adversas.
La frecuencia de las reacciones adversas señaladas a continuación incluye reacciones informadas en pacientes tratados con Rapamune®. En general, los eventos adversos relacionados con la administración de Rapamune® dependieron de la dosis/concentración. Las reacciones adversas se clasifican según las siguientes categorías de frecuencia de CIOMS:
Rapamune® después de la suspensión de la CsA: Se determinó la incidencia de reacciones adversas durante 60 meses en un estudio multicéntrico, con asignación aleatoria y controlado en el que 215 receptores de trasplante renal recibieron Rapamune® como régimen de mantención luego del retiro de la CsA y 215 pacientes recibieron Rapamune con CsA. Todos los pacientes recibieron corticoides. El perfil de seguridad antes de la aleatorización (comienzo de la suspensión de la CsA) fue similar al del grupo de 2 mg de Rapamune® en los estudios de Rapamune® en combinación con CsA. Después de la aleatorización (a los 3 meses) los pacientes del grupo en el que se eliminó la CsA del tratamiento presentaron una incidencia significativamente mayor de elevación de AST/SGOT y ALT/SGPT, daño hepático hipopotasemia, trombocitopenia, cicatrización anormal, acné, íleo y trastornos articulares. Por el contrario, la incidencia de acidosis, hipertensión, toxicidad por la CsA, creatinina aumentada, anormalidades en la función renal, nefropatía tóxica, edema, hiperuricemia, gota neoplasia benigna de piel e hiperplasia gingival fue significativamente mayor en los pacientes que continuaron recibiendo CsA que en los que se eliminó la ciclosporina del tratamiento. La presión media sistólica y diastólica mejoró significativamente tras la suspensión de la CsA. Luego de la suspensión de la CsA (a los 60 meses), la incidencia de infección por Herpes zóster fue significativamente inferior en los pacientes que recibieron Rapamune® luego del retiro de la CsA que en los pacientes que continuaron recibiendo Rapamune® y CsA. La siguiente tabla presenta la incidencia de tumores malignos luego de la suspensión de la CsA. La incidencia de linforma/enfermedad linfoproliferativa fue similar en todos los grupos de tratamiento. La incidencia global de tumores malignos, basada en la cantidad de pacientes que presentaban uno o más carcinomas, fue inferior en los pacientes a los que se les retiró la CsA que en los pacientes que recibieron Rapamune más CsA (10,7% versus 15,8%, respectivamente).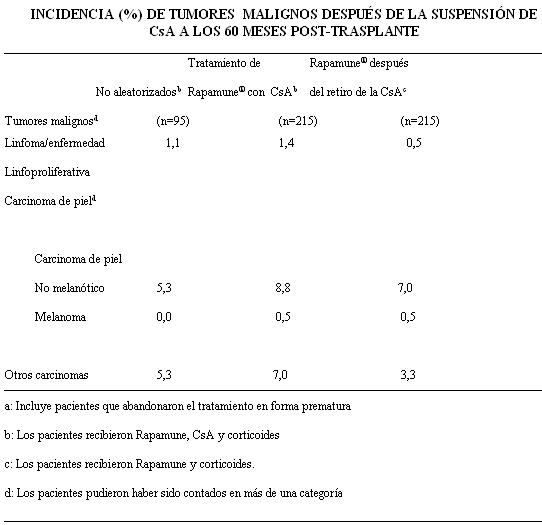
A los 60 meses, la incidencia de carcinomas no cutáneos (linfoma/enfermedad linfoproliferativa más otros carcinomas de la tabla precedente) fue significativamente superior en los cohortes que continuaron con CsA que en los cohortes a los que se les suspendió la CsA (8,4% vs. 3,8% respectivamente). En cáncer de piel, el tiempo promedio hasta la primera aparición estuvo significativamente retardado (491 vs. 1126 días) y al tomar en cuenta que un paciente puede tener múltiples carcinomas de piel, el riesgo relativo (RR = 0,346) de desarrollar cáncer de piel fue significativamente más bajo en el grupo de retiro de la CsA que en el grupo que continuó con la CsA. Se evaluó la seguridad en un estudio clínico controlado que involucraba a 448 pacientes que recibieron, al menos, una dosis del fármaco de estudio (población de seguridad): 224 pacientes recibieron, al menos, una dosis de sirolimus con tacrolimus, y 224 pacientes recibieron, al menos, una dosis de sirolimus con ciclosporina. En general, la incidencia y la naturaleza de los acontecimientos adversos fueron similares a los observados en estudios anteriores combinados con Rapamune. La diarrea y el herpes simplex fueron significativamente más frecuentes en los pacientes que recibieron sirolimus y tacrolimus, mientras que hipertensión, cardiomegalia, linfocele, aumento de la creatinina, acné, trastorno del tracto urinario, quistes ováricos y la toxicidad del inhibidor de la calcineurina ocurrieron con una frecuencia significativamente mayor en los pacientes que recibieron sirolimus y ciclosporina. La incidencia de los tumores malignos fue baja (1,3% en cada grupo). Se evaluó la seguridad en un estudio clínico controlado en pacientes pediátricos ( < 18 años) con trasplante renal considerados de alto riesgo inmunológico, definido como antecedentes de uno o más episodios de rechazo agudo y/o presencia de nefropatía crónica por aloinjerto en una biopsia renal. Se asoció el empleo de Rapamune en combinación con inhibidores de la calcineurina y corticosteroides con un mayor riesgo de deterioro de la función renal, anormalidades en lípidos séricos (que incluyeron pero sin limitarse a aumento de colesterol y triglicéridos séricos) e infecciones urinarias. No se ha establecido la seguridad y eficacia de la conversión de inhibidores de la calcineurina a Rapamune® en la mantención de pacientes con trasplante renal. En un estudio en curso que evalúa la seguridad y eficacia de la conversión de inhibidores de la calcineurina a Rapamune® (niveles deseados de sirolimus de 12-20 ng/ml por ensayo cromatográfico) en la mantención de pacientes con trasplante renal, se suspendió la incorporación en el subgrupo de pacientes (n=90) con índice de filtración glomerular basal inferior a 40 mL/min. Se observó una mayor incidencia de eventos adversos serios, tales como neumonía, rechazo agudo, pérdida del injerto y muerte en este brazo de tratamiento con Rapamune® (n=60, tiempo promedio postrasplante 36 meses). La administración concomitante de sirolimus con un inhibidor de la calcineurina puede aumentar el riesgo de síndrome hemolítico urémico /púrpura trombocitopénica trombótica/microangiopatía trombótica (SHU/PTT/MAT) inducidos por los inhibidores de la calcineurina. En pacientes con función de injerto demorada, Rapamune puede demorar en recuperar la función renal. Enfermedad pulmonar intersticial: Se han comunicado casos de enfermedad pulmonar intersticial [que incluyeron neumonitis y, con poca frecuencia, bronquiolitis obliterante con neumonía organizante y fibrosis pulmonar], en ocasiones fatales, sin identificación de la etiología infecciosa en pacientes que recibieron regímenes inmunosupresores entre ellos Rapamune®. En algunos casos, la enfermedad pulmonar intersticial se resolvió al suspender el tratamiento o reducir la dosis de Rapamune®. El riesgo puede ser mayor a medida que aumenta el nivel mínimo de sirolimus. Infecciones virales latentes: Se ha observado nefropatía y leucoencefalopatía multifocal progresiva asociada al virus BK y JC en pacientes que recibian inmunosupresores, incluyendo Rapamune. Estas infecciónes pueden estar asociadas con resultados serios, incluyendo pérdida del injerto renal. Hepatotoxicidad: Se ha informado de hepatotoxicidad, incluyendo casos fatales de necrosis hepática con niveles mínimos de sirolimus elevados (es decir, por encima de los niveles terapéuticos). Cicatrización anormal: Se ha informado de cicatrización anormal después de la cirugía de trasplante, incluyendo dehiscencia facial, hernia incisional y disrupción de la anastomosis (por ejemplo, herida, vascular, vía aérea, ureteral, biliar). Otras experiencias clínicas: Se ha informado Azoospermia con el uso de Rapamune y en la mayoría de los casos ha sido reversible después de la discontinuación de Rapamune. Se han reportado casos de enterocolitis por clostridium difficile en pacientes que reciben sirolimus.
Precauciones.
Cicatrización de heridas y Acumulación de Fluidos: Los inhibidores mTOR (mammalian Target Of Rapamycin - blanco de rapamicina en mamíferos) tales como el Sirolimus han mostrado in vitro la inhibición de la producción de ciertos factores del crecimiento que pueden afectar la angiogénesis, la proliferación de fibroblastos y la permeabilidad vascular. Se ha reportado deterioro o retardo en la cicatrización en pacientes recibiendo Rapamune, incluyendo linfocele y dehiscencia de heridas. Linfocele, una complicación quirúrgica conocida del transplante renal, ocurrió con una frecuencia significativamente mayor en pacientes tratados con Rapamune® Deben considerarse medidas postquirúrgicas apropiadas para minimizar esta complicación. Pacientes con un BMI mayor que 30 Kg/m2 pueden estar expuestos a un mayor riesgo de cicatrización anormal, basado en la información de la literatura médica. También se ha reportado acumulación de fluidos, incluyendo edema periférico, linfedema, derrame pleural y derrames pericardiales (incluyendo derrames hemodinámicamente significantes en niños y adultos), en pacientes recibiendo Rapamune. Carcinomas de piel: La inmunosupresión aumenta el riesgo de desarrollo de linfomas y otros procesos malignos, particularmente de piel. Por lo tanto, los pacientes que tomen Rapamune® deberían limitar la exposición al sol y rayos UV mediante el empleo de ropa protectora y una crema pantalla solar con alto factor de protección. Hiperlipidemia: El empleo de Rapamune® puede aumentar los triglicéridos y el colesterol sérico, necesitando tratamiento en algunos casos. Los pacientes deberán ser controlados para detección de hiperlipidemia. Rabdomiólisis: En los estudios clínicos llevados a cabo, la administración concomitante de Rapamune® e inhibidores de la HMG CoA reductasa y/o fibratos fue bien tolerada. Durante el tratamiento con Rapamune® CsA, deberá controlarse a los pacientes que reciban un inhibidor de la HMG CoA reductasa y/o fibratos por posible desarrollo de rabdomiólisis y otros efectos adversos descritos en los respectivos prospectos de estos agentes. Función renal: Los pacientes tratados con CsA y Rapamune presentaron niveles más altos de creatinina sérica e índices más bajos de filtración glomerular en comparación con los pacientes del grupo de control tratados con CsA y placebo o azatioprina. La declinación de la función renal fue mayor en los pacientes que recibieron Rapamune y CsA en comparación con los tratamientos control. Por lo tanto, se deberá controlar la función renal durante la coadministración de Rapamune con ciclosporina y durante la coadministración de Rapamune con tacrolimus. En pacientes con niveles elevados de creatinina sérica se recomienda considerar ajustes adecuados del régimen inmunosupresor, incluso interrupción de Rapamune y/o ciclosporina y/o tacrolimus. En un estudio que comparó un régimen de Rapamune® y CsA contra uno en el que se retiraba la CsA 2-4 meses después del trasplante, los pacientes a los que se les suspendió la CsA presentaron niveles de creatinina sérica significativamente superiores e índices de filtración glomerular significativamente inferiores a partir de los 12 meses hasta los 60 meses, y sobrevida del injerto significativamente inferior a los 48 meses, en cuyo momento el patrocinador decidió retirar a los pacientes asignados al brazo de tratamiento de Rapamune y CsA. Cuando se modificó el protocolo, todos los pacientes habían llegado a los 48 meses y algunos completaron los 60 meses del estudio. En pacientes con riesgo inmunológico reducido a moderado, deberá considerarse la continuación del tratamiento combinado con CsA después de 4 meses del trasplante sólo cuando los beneficios superen los riesgos de dicha combinación en cada caso en particular. En pacientes con retraso en la función de injerto, Rapamune puede demorar la recuperación de la función renal. Proteinuria: Se recomienda un monitoreo cuantitativo periódico de la excreción urinaria de proteínas. En un estudio evaluando la conversión de inhibidores de calcineurina (CNI) a Rapamune en mantención en pacientes con trasplante renal de 6 a 120 meses después del trasplante, un aumento de la excreción urinaria de proteinas fue observada comúnmente de 6 a 24 meses después de la conversión a Rapamune comparada con la continuación de inhibidores de calcineurina (CNI) (23,6% vs 12,8%, respectivamente) [ver Reacciones Adversas y Eficacia Clínica]. Aquellos pacientes en el cuartil más alto de excreción urinaria de proteínas antes de la conversión a Rapamune (proporción proteína urinaria a creatinina ≥0,27) fueron aquellos cuya excreción de proteínas aumentó más después de la conversión. También en este estudio se informó principio de nefrosis (síndrome nefrótico) en el 2% de los pacientes. Se observó una reducción en el grado de excreción urinaria de proteínas en pacientes individuales luego de la discontinuación de Rapamune. No se ha establecido la seguridad y eficacia de la conversión de inhibidores de calcineurina a sirolimus en la población con trasplante renal en mantención. Conversión a Rapamune en pacientes con tasa de Filtración Glomerular < 40 mL/min: En un estudio evaluando la conversión de inhibidores de calcineurina (CNI) a Rapamune en mantención en pacientes con trasplante renal 6-120 meses después del trasplante (ver Eficacia Clínica), en un estrato de la rama de tratamiento de Rapamune con una tasa de filtración glomerular calculada de menos de 40 mL/min, hubo una mayor tasa de eventos adversos serios, incluyendo neumonía, rechazo agudo, pérdida del injerto y muerte. No ha sido establecida la seguridad y eficacia de la conversión de inhibidores de calcineurina a Rapamune en mantención en pacientes con trasplante renal. Uso de novo sin un inhibidor de calcineurina (CNI): No está establecida la seguridad y eficacia del uso de novo de Rapamune® sin un inhibidor de calcineurina (CNI) en pacientes con trasplante renal. En dos estudios clínicos multicéntricos, los pacientes de trasplante renal de novo tratados con Rapamune, MMF, esteroides y un antagonista de receptor IL-2 hubo una tasa significativamente mayor de rechazos y una tasa numéricamente más alta de muertes comparado con pacientes tratados con inhibidores de calcineurina, MMF, esteroides y antagonista de receptor IL-2. Un beneficio, en términos de una mejor función renal, no fue aparente en las ramas de tratamiento con uso de novo de Rapamune sin un inhibidor de calcineurina. Se debería notar que se usó un esquema abreviado de administración de daclizumab en uno de los estudios. Síndrome hemolítico urémico /púrpura trombocitopénica trombótica/microangiopatía trombótica (SHU/PTT/MAT) inducidos por inhibidores de la calcineurina: La administración concomitante de sirolimus y un inhibidor de la calcineurina puede aumentar el riesgo de (SHU/PTT/MAT) inducidos por los inhibidores de la calcineurina. Empleo concomitante de inhibidores de la enzima convertidora de angiotensina (ECA): En raros casos, la administración concomitante de sirolimus con inhibidores ECA ha provocado reacciones del tipo edema angioneurótico. Enfermedad pulmonar intersticial: Se han comunicado casos de enfermedad pulmonar intersticial (que incluyeron neumonitis y, con poca frecuencia, bronquitis obliterante con neumonía organizante y fibrosis pulmonar), en ocasiones fatales, sin identificación de la etiología infecciosa en pacientes que recibieron regímenes inmunosupresores, entre ellos Rapamune®. En algunos casos, la enfermedad pulmonar intersticial se resolvió al suspender el tratamiento o reducir la dosis de Rapamune®. El riesgo puede ser mayor a medida que aumenta el nivel mínimo de sirolimus. Infecciones virales latentes: Los pacientes tratados con inmunosupresores, incluyendo Rapamune, están expuestos a un mayor riesgo de infecciones oportunistas, incluyendo la activación de infecciones virales latentes. Entre estas condiciones está la nefropatía asociada al virus BKy la leucoencefalopatía multifocal progresiva (LMP) asociada al virus JC. Estas infecciónes están a menundo relacionadas a una elevada carga inmunosupresora total y pueden derivar en resultados graves o fatales, incluyendo pérdida del injerto. Los médicos deberían considerar las infecciones virales latentes en los diagnósticos diferenciales en pacientes inmunodeprimidos con deterioro en la función renal. Profilaxis antimicrobiana: Profilaxis antimicrobiana por Pneumocystis carinii deberá administrarse durante 1 años después del transplante. Se recomienda profilaxis frente al citomegalovirus (CMV) por 3 meses después del trasplante, particularmente en pacientes con mayor riesgo de contraer infección por CMV. Anticoncepción: Se deberá iniciar una anticoncepción eficaz antes del tratamiento con Rapamune® y mantenerla durante toda la terapia y durante las 12 semanas posteriores a la interrupción del fármaco. Empleo en pacientes de alto riesgo: No se ha evaluado en forma adecuada la seguridad y eficacia del retiro de la CsA en pacientes con trasplante renal de alto riesgo y por lo tanto no se recomienda dicho uso. Este grupo incluye a pacientes con rechazo agudo grado III de Banff93 o rechazo vascular antes del retiro de la CsA, pacientes dializados o aquellos con creatinina sérica > 4,5 mg/dl, pacientes de raza negra, pacientes con retrasplante renal, con multitrasplante de órganos y pacientes con alto panel de anticuerpos reactivos. Embarazo: No se han llevado a cabo estudios en mujeres embarazadas. En estudios con animales la toxicidad embrio /fetal se manifestó como mortalidad y pesos fetales reducidos (con retrasos asociados en la osificación del esqueleto). Rapamune® debe ser empleado durante el embarazo solamente si el beneficio sobrepasa el riesgo potencial para el embrión /feto. Lactancia: Sirolimus se excreta en mínima cantidad en la leche de ratas que lactan. No se sabe si sirolimus se excreta en la leche humana debe tomarse una decisión sobre si interrumpir la lactancia o interrumpir el tratamiento con Rapamune. Uso pediátrico: No se ha establecido la seguridad y eficacia de Rapamune® en pacientes pediátricos menores de 13 años. Si se emplea en niños < 13 años, se recomienda controlar los niveles mínimos de sirolimus en sangre. En pacientes ≥13 años que pesen menos de 40 Kg., la dosis inicial de carga debería ser de 3 mg/m2. La dosis de mantención deberá ajustarse según el área de superficie corporal a 1 mg/m2/día. Uso geriátrico: Los estudios clínicos de Rapamune® no incluyeron un número suficiente de pacientes con edades de 65 años o más para determinar si la seguridad y eficacia difieren en esta población respecto de las de pacientes más jóvenes. Los datos sobre la concentración mínima de sirolimus en 35 pacientes renales trasplantados > 65 años fueron similares a los de la población adulta (n=822) de 18 a 65 años. Pacientes con daño hepático: En pacientes con disfunción hepática, se recomienda reducir la dosis de mantención de Rapamune en aproximadamente un tercio. No es necesario modificar la dosis de carga de Rapamune® sirolimus. No se ha estudiado la farmacocinética del sirolimus en pacientes con disfunción hepática severa. En pacientes con deterioro hepático, se recomienda controlar los niveles de sirolimus en sangre. Pacientes con daño renal: En base a datos farmacocinéticos clínicos, la dosis de Rapamune® sirolimus no requiere ser ajustada en casos de deterioro de la función renal. Monitoreo de los niveles sanguíneos de Sirolimus: Los niveles sanguíneos de sirolimus deben monitorearse en los siguientes casos: pacientes recibiendo Rapamune® en concentración controlada. Pacientes pediátricos. Pacientes con deterioro hepático. Durante la administración concurrente de inductores e inhibidores fuertes de CYP3A4 y de la P-glucoproteína (P-gp). Si la dosificación de ciclosporina es marcadamente reducida o interrumpida. Se recomienda que a los pacientes que se cambien de la formulación en solución oral a las grageas, sobre la base de mg por mg, se les realice un monitoreo de contenido sanguíneo y niveles plasmáticos, tomado 1 a 2 semanas después del cambio de las formulaciones, para confirmar que la concentración de sirolimus está dentro de los rangos deseados. El monitoreo de los niveles terapéuticos de la droga no debe constituir la base exclusiva para ajustar el tratamiento con Sirolimus. Deberá prestarse especial atención a los signos/síntomas, biopsias de tejido y parámetros de laboratorio. En los estudios clínicos controlados con CsA concomitante, los niveles medios mínimos de sirolimus a los 6 meses después del trasplante, expresados como valores de ensayo cromatográfico fueron de 7,2 ng/mL (rango -3,6 - 11 ng/mL percentilo 10° a 90°) para el grupo de tratamiento con 2 mg/día (n=226), y de 14 ng/mL (rango -8,0-22 ng/mL percentilo 10° a 90°) para la dosis de 5 mg/día (n=219; valores obtenidos por inmunoensayo experimental, pero expresados como valores cromatográfricos equivalentes, empleando un sesgo +20% para el inmunoensayo). En los estudios clínicos controlados con retiro de CsA, las concentraciones sanguíneas medias mínimas de sirolimus durante el 4° a 12° mes después del trasplante, medidas por cromatografía, fueron de 8,6 ng/mL (rango 5,0 - 12,7 ng/mL [percentilo 10° a 90°] en el grupo de tratamiento concomitante de Rapamune y Ciclosporina (n=205) y fueron 18,6 ng/mL (rango 13,6 - 22,4 ng/mL [percentilo 10° a 90°]) en el grupo de retiro de la Ciclosporina (n=201). Al mes 60, las concentraciones sanguíneas medias mínimas de sirolimus se mantuvieron estables en el grupo de tratamiento concomitante de Rapamune y Ciclosporina (n=71) a 9,1 ng/mL (rango 5,4 a 13,9 ng/mL [percentilo 10° a 90°]). En el grupo de retiro de Ciclosporina (n=104) al mes 60 la concentración sanguínea media de sirolimus descendió a 16,3 ng/mL (rango 11,2 a 21,9 ng/mL [percentilo 10° a 90°]). En los estudios clínicos de concentración controlada en pacientes adultos de alto riesgo, las concentraciones sanguíneas medias mínimas, durante el 9° a 12° meses después del trasplante, medidas por cromatografía, en el grupo de sirolimus/tacrolimus, fueron 10,7 ng/mL (rango 5,6 - 15,1 ng/mL [percentilo 10° a 90°]) (n=117) y las concentraciones sanguíneas medias mínimas de tacrolimus fueron de 5,3 ng/mL (rango 3,0 - 8,6 ng/mL [percentilo 10° a 90°]). Adicionalmente, las concentraciones sanguíneas medias mínimas de sirolimus en el grupo de sirolimus/ciclosporina fueron 11,2 ng/mL (rango 6,8 - 15,9 ng/mL [percentilo 10° a 90°]) (n=127), y las concentraciones sanguíneas medias mínimas de ciclosporina fueron 133 ng/mL (rango 54 - 215 ng/mL [percentilo 10° a 90°]). Métodos de ensayo: Los rangos recomendados de concentración mínima de 24 horas para sirolimus se basan en métodos cromatográficos. Se han utilizado distintos métodos de ensayo para medir las concentraciones sanguíneas de sirolimus. En la práctica clínica actual, las concentraciones sanguíneas de sirolimus se determinan por cromatografía y por inmunoensayo. Los valores de la concentración obtenidos por estos métodos diferentes no son recíprocos. Deberán realizarse ajustes al rango objetivo de acuer