RECORMON® Solución Inyectable 30.000 UI/0,6 mL
ROCHE
Antianémico.
Composición.
Principio activo: epoetina beta (eritropoyetina humana recombinante, producida mediante tecnología de ADN recombinante en la línea celular CHO). Recormon se presenta en forma de liofilizado y disolvente para solución inyectable, en forma de liofilizado y disolvente para solución inyectable en cartuchos y en forma de solución inyectable en jeringas precargadas. El producto reconstituido es una solución incolora, límpida o ligeramente opalescente. Solución inyectable en jeringas precargadas: 1.000UI = 8,3 microgramos de epoetina beta con 0,3ml de solución inyectable; 2.000UI = 16,6 microgramos de epoetina beta con 0,3ml de solución inyectable; 30.000UI = 250 microgramos de epoetina beta con 0,6ml de solución inyectable. Excipientes: todas las presentaciones contienen fenilalanina (hasta 5,0mg por vial multidosis, 0,5mg por cartucho y 0,3mg por jeringa precargada) (ver Advertencias).
Farmacología.
La eficacia biológica de la epoetina tras su administración por vía intravenosa y subcutánea se ha demostrado en diversos modelos animales in vivo (ratas normales y urémicas, ratones policitémicos, perros). Después de recibir epoetina beta, aumentaban los recuentos de eritrocitos y reticulocitos, los valores de hemoglobina y la incorporación globular de Fe59. In vitro (cultivos de células esplénicas de ratón) se ha observado un aumento de la incorporación de H3-timidina a las células esplénicas nucleadas eritroides tras la incubación con epoetina beta. Por otro lado, en cultivos de células de médula ósea humana se ha puesto de manifiesto que la epoetina beta estimula específicamente la eritropoyesis y que no afecta a la leucopoyesis. No se han detectado efectos citotóxicos de la epoetina beta sobre la médula ósea o la piel del ser humano. Tras la administración de una dosis única de epoetina beta, no se ha apreciado ningún cambio del comportamiento y la actividad locomotora del ratón, ni de la función respiratoria del perro. Propiedades farmacodinámicas: en su composición de aminoácidos y carbohidratos, la epoetina beta es idéntica a la eritropoyetina aislada de la orina de pacientes anémicos. La eritropoyetina es una glicoproteína que estimula la formación de eritrocitos a partir de los progenitores eritroides. Actúa como factor estimulante de la mitosis y hormona de diferenciación. Mecanismo de acción: la eritropoyetina es una glicoproteína que, como factor de crecimiento, estimula fundamentalmente la formación de eritrocitos a partir de los progenitores eritroides. Actúa como factor estimulante de la mitosis y hormona de diferenciación. Ensayos clínicos/eficacia: en este apartado se describen los estudios controlados y aleatorizados recientemente completados con epoetina beta en pacientes con anemia renal o pacientes con cáncer tratados con quimioterapia/radioterapia. Pacientes con anemia por insuficiencia renal crónica: se llevó a cabo un estudio aleatorizado abierto con epoetina beta en 605 pacientes en prediálisis (estudio CREATE) que presentaban anemia leve o moderada (concentración de Hb: 11-12,5g/dl). El objetivo principal era investigar si una corrección de la hemoglobina con valores altos (13-15g/dl) reduciría la morbilidad cardiovascular (CV) en comparación con el tratamiento estándar de la anemia (con un valor diana de Hb de 10,5-11,5g/dl). No se observó beneficio alguno con una corrección de Hb con valores altos en comparación con la corrección estándar de la anemia. Por el contrario, se observaron menos episodios en el grupo de tratamiento estándar (47 frente a 58 episodios, RR 0,78; p = 0,20). Se observó una diferencia en el tiempo transcurrido hasta el inicio de la diálisis, favorable al grupo de corrección estándar de la anemia (111 y 127 episodios, mediana del tiempo hasta la diálisis de 41 meses y 36 meses, prueba de rangos logarítmicos p = 0,034, respectivamente), aunque no hubo diferencias en la mediana del aclaramiento de creatinina a lo largo del tiempo entre los dos grupos en estudio. La calidad de vida (evaluada mediante el cuestionario de salud SF-36) había mejorado significativamente al cabo de 1 año (p = 0,003) en el grupo con un valor diana de Hb alto. En otro estudio aleatorizado abierto, llevado a cabo en 172 pacientes con nefropatía diabética inicial (estudio ACORD), se investigó el efecto de una corrección de Hb con valores altos (valor diana de Hb de 13-15g/dl) y una corrección de Hb estándar (valor diana de Hb de 10,5-11,5g/dl) sobre la estructura y la función cardíacas. Al final del estudio no había diferencias significativas entre ambos grupos en la variable de valoración principal: el índice de masa ventricular izquierda ajustado respecto del valor basal (p = 0,88). No había diferencias estadísticamente significativas entre los grupos de tratamiento en cuanto a cambio del valor basal de aclaramiento de la creatinina calculado, tiempo hasta el aumento al doble de la creatinina sérica o un análisis de los pacientes con progresión rápida. La puntuación de salud general según el cuestionario de valoración de la calidad de vida (SF-36) mejoró significativamente (p = 0,04) en el grupo con un valor diana de Hb alto. Pacientes con cáncer y anemia sintomática tratados con quimioterapia: en un estudio controlado con placebo en el que se utilizó epoetina beta en 351 pacientes con cáncer de cabeza y cuello (estudio ENHANCE), se administró el fármaco para mantener la concentración de hemoglobina en 14g/dl en las mujeres y en 15g/dl en los varones. La supervivencia sin progresión locorregional fue significativamente inferior en los pacientes tratados con epoetina beta (RR = 1,62, p = 0,0008). En los resultados y la interpretación de este estudio influyó la confusión introducida por los desequilibrios existentes entre los grupos de tratamiento, en especial en cuanto a la localización tumoral, el tabaquismo y la heterogeneidad de la población en estudio. Un estudio controlado, aleatorizado y abierto, en el que se utilizó epoetina beta en 463 pacientes con cáncer de mama metastásico tratadas con quimioterapia (BRAVE) y que se había diseñado para identificar una posible mejora significativa de la supervivencia, no mostró ninguna diferencia significativa entre los grupos de control y de epoetina beta en cuanto a supervivencia global (p = 0,52) o tiempo hasta la progresión tumoral (p = 0,45). Hubo un mayor número de pacientes del grupo de control (64/232; 27,6%) que del grupo de epoetina beta (40/231; 17,3%) que fueron tratadas con transfusiones o presentaron episodios de anemia grave (p = 0,009), lo que refleja la eficacia del tratamiento con epoetina beta para prevenir las transfusiones mediante un aumento efectivo de la hemoglobina. Hubo un mayor porcentaje de pacientes tratadas con epoetina beta que experimentaron episodios tromboembólicos (ETE) durante el estudio que en el grupo de control (13% frente a 6%), y el tiempo transcurrido hasta los ETE fue inferior en el grupo de epoetina beta que en el de control (p = 0,008). Sin embargo, el porcentaje de pacientes que sufrieron ETE graves (3% en el grupo de control frente a 4% en el de epoetina beta) o ETE letales (2% en cada grupo) fue comparable. En un estudio controlado, aleatorizado y abierto, en el que se utilizó epoetina beta en 74 pacientes con cáncer de cuello uterino tratadas con radioquimioterapia (estudio MARCH), no se observó una correlación entre el aumento de la hemoglobina y la reducción del número de fracasos terapéuticos (respuesta a la radioquimioterapia). En consecuencia, se decidió no continuar con la segunda fase del estudio. Se llevó a cabo un metanálisis con la inclusión de todos los estudios clínicos controlados en pacientes con cáncer y anemia tratados con epoetina beta (12 estudios con un total de 2.301 pacientes). Los resultados de este metanálisis confirman la eficacia conocida de epoetina beta para elevar las concentraciones de hemoglobina y reducir el riesgo de necesitar transfusiones de sangre. En el conjunto de la población, incluidos también a pacientes con valores iniciales de Hb de hasta 13g/dl, no se observó un aumento significativo del riesgo de muerte en el grupo de epoetina beta en comparación con el grupo de control (RR: 1,13, IC del 95% 0,87 a 1,46, p = 0,34). En los pacientes con una hemoglobina basal ≤11g/dl, la RR para la supervivencia global fue de 1,09 (IC del 95% 0,80 a 1,47, p = 0,58). Por lo que respecta al tiempo transcurrido hasta la progresión de la enfermedad, la RR fue de 0,85 (IC del 95%: 0,72 a 1,01, p = 0,07) en la población global de pacientes. Al limitar el análisis a los pacientes con una hemoglobina basal ≤11g/dl, la RR era de 0,80 (IC del 95% 0,65 a 0,99, p = 0,04). Este metanálisis confirmó también el aumento de la tasa de episodios tromboembólicos (ETE) registrados (ver Ensayos clínicos), con una tasa de ETE del 7% en el grupo de epoetina beta, frente al 4% en el grupo de control. Propiedades farmacocinéticas: según los estudios farmacocinéticos en voluntarios sanos y pacientes urémicos, la semivida de la epoetina beta administrada por vía intravenosa oscila entre 4 y 12 horas, y el volumen de distribución es entre una y dos veces superior al volumen plasmático. Se han obtenido resultados similares en los ensayos con ratas urémicas y normales. Absorción: tras la administración subcutánea de epoetina beta a pacientes urémicos, su absorción prolongada se traduce en una concentración sérica en equilibrio cuyo valor máximo se alcanza al cabo de 12 a 28 horas. La biodisponibilidad de la epoetina beta por vía subcutánea varía entre el 23% y el 42% de la alcanzada por vía intravenosa. Distribución: según los estudios farmacocinéticos en voluntarios sanos y pacientes urémicos, el volumen de distribución es entre una y dos veces superior al volumen plasmático. Eliminación: según los estudios farmacocinéticos en voluntarios sanos y pacientes urémicos, la semivida de la epoetina beta administrada por vía intravenosa oscila entre 4 y 12 horas. Tras la administración subcutánea de epoetina beta a pacientes urémicos, la semivida terminal es más alta que la observada tras la administración intravenosa y se sitúa entre 13 y 28 horas. Farmacocinética en poblaciones especiales: no se han realizado estudios formales del efecto de la insuficiencia hepática sobre la farmacocinética de la epoetina beta. Datos preclínicos sobre seguridad: carcinogenicidad: un estudio de carcinogenicidad con eritropoyetina homóloga en ratones no reveló ningún signo de potencial proliferativo o carcinogénico. Otros efectos: los datos preclínicos no ponen de manifiesto ningún riesgo especial para el ser humano según los resultados de estudios convencionales de tóxico-farmacología, toxicidad en dosis repetidas, genotoxicidad y toxicidad en la reproducción.
Indicaciones.
Tratamiento de la anemia sintomática asociada a la insuficiencia renal crónica (IRC) en pacientes dializados. Tratamiento de la anemia renal sintomática en pacientes todavía no dializados. Prevención de la anemia de la prematuridad en recién nacidos con un peso al nacer de 750 a 1.500 g y una edad de gestación inferior a 34 semanas. Tratamiento de la anemia sintomática en pacientes adultos con mieloma múltiple, linfoma no Hodgkin de bajo grado o leucemia linfocítica crónica, que tienen deficiencia relativa de eritropoyetina y están recibiendo terapia antitumoral. La deficiencia viene definida como un nivel bajo de eritropoyetina en suero, inadecuado en relación al grado de anemia. Aumento de la producción de sangre autóloga en pacientes participantes en un programa de predonación. Su uso en esta indicación debe sopesarse con el aumento descrito del riesgo de episodios tromboembólicos. En los pacientes con anemia moderada (Hb 10-13g/dl [6,21 - 8,07mmol/l], sin ferropenia) sólo se aplicará este tratamiento si no se dispone de sistemas para la conservación de sangre o éstos son insuficientes ante una intervención de cirugía mayor programada que requiera un gran volumen de sangre (4 unidades o más de sangre en las mujeres o 5 unidades o más en los varones).
Dosificación.
El tratamiento con Recormon debe ser instaurado por médicos con experiencia en las indicaciones antes citadas. Dado que ha habido casos aislados de reacciones anafilactoides, se recomienda administrar la primera dosis bajo supervisión médica. Solución inyectable en jeringas precargadas: las jeringas precargadas de Recormon están listas para el uso. Bajo ninguna circunstancia debe administrarse más de una dosis por jeringa; el producto es únicamente para un solo uso. Tratamiento de pacientes anémicos con insuficiencia renal crónica: la solución reconstituida puede administrarse por vía subcutánea o intravenosa. Si la vía elegida es la intravenosa, se inyectará la solución a lo largo de 2 minutos, aproximadamente; por ejemplo a los pacientes hemodializados a través de la fístula arteriovenosa al final de la diálisis. En los pacientes no hemodializados debe preferirse siempre la administración subcutánea, con objeto de evitar la punción de venas periféricas. En los pacientes con IRC, el objetivo del tratamiento es alcanzar un valor de Hb de 10-12g/dl. No debe superarse una cifra de Hb de 12g/dl. Si el aumento de la hemoglobina es superior a 2g/dl (1,3mmol/l) en 4 semanas, deberá contemplarse una reducción apropiada de la dosis. En presencia de hipertensión o de enfermedades cardiovasculares, cerebrovasculares o vasculares periféricas ya existentes, el aumento semanal de Hb y el valor diana de Hb deberán determinarse de forma individualizada, teniendo en cuenta el cuadro clínico. Debe mantenerse una vigilancia estrecha de los pacientes para garantizar la utilización de la dosis más baja posible de Recormon que proporcione un control adecuado de los síntomas de anemia. El tratamiento con Recormon consta de dos fases. 1. Fase de corrección: administración subcutánea (todas las formas farmacéuticas): la dosis inicial es de 3 x 20UI/kg de peso por semana. Puede aumentarse la dosis cada 4 semanas en 3 x 20UI/kg por semana si el incremento de la Hb no es suficiente ( < 0,25g/dl por semana). La dosis semanal puede fraccionarse en dosis diarias. Administración intravenosa (polvo y disolvente para solución inyectable y jeringas precargadas únicamente): la dosis inicial es de 3 x 40UI/kg por semana. Al cabo de 4 semanas, puede aumentarse la dosis a 80UI/kg (tres veces por semana). Si es preciso aumentarla de nuevo, deberá hacerse a razón de 20UI/kg tres veces por semana, a intervalos mensuales. La dosis máxima no debe exceder de 720UI/kg/semana por ninguna de ambas vías de administración. 2. Fase de mantenimiento: para mantener el valor diana de Hb de aproximadamente 10-12g/dl, primero debe reducirse la dosis a la mitad de la previamente administrada. A continuación, se ajustará la dosis a intervalos de dos a cuatro semanas, de manera individualizada en cada paciente (dosis de mantenimiento). Si la administración es subcutánea, la dosis semanal puede administrarse en una inyección o fraccionarse en tres o siete dosis a la semana. Los pacientes que permanezcan estables con el régimen de una sola dosis semanal pueden pasar a una sola administración cada dos semanas. En este caso, puede ser necesario un aumento de la dosis. Por lo general, el tratamiento con Recormon es de larga duración. Ahora bien, se lo puede interrumpir en cualquier momento en caso de necesidad. Los datos sobre la pauta de una vez a la semana se han obtenido de estudios clínicos con una duración del tratamiento de 24 semanas. Tratamiento de la anemia en pacientes con mieloma múltiple, linfoma no Hodgkin de bajo grado o leucemia linfocítica crónica que reciben quimioterapia: la solución reconstituida se administra por vía subcutánea; la dosis semanal puede administrarse en una única inyección o fraccionarse en 3-7 dosis semanales. La dosis inicial recomendada es de 30.000UI por semana (equivalentes a aproximadamente 450UI/kg de peso por semana en un paciente de peso medio). El tratamiento con Recormon está indicado cuando el valor de hemoglobina es ≤11g/dl (6,83mmol/l). El valor de hemoglobina no debe exceder de 13g/dl (8,07mmol/l) (ver Estudios clínicos/eficacia). Si al cabo de 4 semanas se ha elevado el valor de hemoglobina por lo menos en 1g/dl (0,62mmol/l), debe mantenerse la dosis utilizada. Si el valor de hemoglobina no se ha elevado por lo menos en 1g/dl (0,62mmol/l), debe considerarse una dosis semanal el doble de alta. Si al cabo de 8 semanas no se ha elevado el valor de hemoglobina por lo menos en 1g/dl (0,62mmol/l), no es probable que haya respuesta y debe suspenderse el tratamiento. El tratamiento debe proseguirse hasta 4 semanas después de finalizada la quimioterapia. La dosis máxima no debe exceder de 60.000UI por semana. Una vez alcanzado el objetivo terapéutico en un paciente determinado, debe reducirse la dosis en un 25-50% para mantener la concentración de hemoglobina en ese nivel. Si es necesario, puede reducirse la dosis aún más con el fin de que la concentración de hemoglobina no supere los 13g/dl. Si el aumento de la concentración de hemoglobina en 4 semanas es superior a 2g/dl (1,3mmol/l), debe reducirse la dosis en un 25-50%. Tratamiento para aumentar la cantidad de sangre autóloga: la solución reconstituida se administra por vía intravenosa, a lo largo de unos 2 minutos, o por vía subcutánea. Recormon se administra dos veces por semana, durante 4 semanas. Cuando el hematócrito del paciente permita la donación de sangre, es decir, cuando sea ≥33%, se administrará Recormon al final de la donación. En ningún momento del tratamiento debe sobrepasarse un hematócrito del 48%. La dosis ha de determinarla el equipo quirúrgico en cada paciente, individualmente, a partir del volumen requerido de sangre predonada y de la reserva endógena de eritrocitos: 1. El volumen necesario de sangre predonada dependerá de la pérdida prevista de sangre, del uso de medios para conservarla y del estado físico del paciente. Este volumen debería ser el que se prevea que será suficiente para evitar transfusiones de sangre homóloga. 2. La cantidad requerida de sangre predonada se expresa en unidades, equivaliendo una unidad del nomograma a 180ml de eritrocitos. 3. La capacidad de donar sangre depende fundamentalmente del volumen sanguíneo del paciente y del hematócrito basal. Una y otra variables determinan la reserva eritrocitaria endógena, la cual puede calcularse mediante la fórmula siguiente: reserva eritrocitaria endógena = volumen sanguíneo (ml) x (hematócrito - 33) ÷ 100. Mujeres: volumen sanguíneo (ml) = 41 (ml/kg) x peso corporal (kg) + 1.200 (ml). Hombres: volumen sanguíneo (ml) = 44 (ml/kg) x peso corporal (kg) + 1.600 (ml) (peso corporal ≥45kg). La indicación para un tratamiento con Recormon y, en su caso, la dosis única deben determinarse en función del volumen requerido de sangre predonada y la reserva endógena de eritrocitos, según los gráficos siguientes: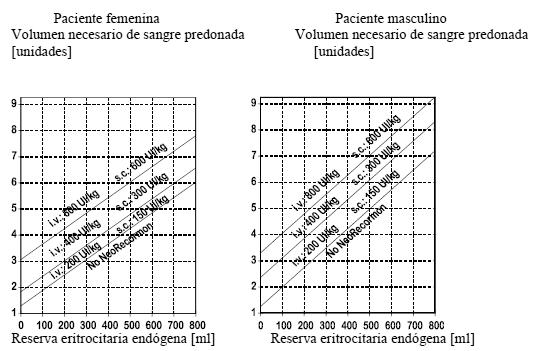
La dosis así determinada se administra dos veces por semana, durante 4 semanas. La dosis máxima no debe exceder de 1.600UI/kg/semana por vía intravenosa o de 1.200UI/kg/semana por vía subcutánea. Prevención de la anemia de la prematuridad: para esta indicación, solamente puede utilizarse la solución inyectable en jeringas precargadas. La solución se administra por vía subcutánea en una dosis de 3 x 250UI/kg de peso por semana. El tratamiento con Recormon debe iniciarse lo antes posible, preferiblemente en los tres primeros días de vida. En los recién nacidos prematuros que hayan recibido una transfusión antes de iniciar el tratamiento con Recormon no es probable que el efecto favorable sea tan alto como el observado en los recién nacidos que no han recibido ninguna transfusión. La duración del tratamiento debe ser de 6 semanas. Pautas posológicas especiales: niños y adolescentes: los resultados de los estudios clínicos realizados en niños y adolescentes muestran que, en general, cuanto menor es la edad del paciente, mayores son las dosis de Recormon necesarias. No obstante, debe seguirse la pauta de administración recomendada, puesto que no es posible predecir la respuesta individual (ver Uso en pediatría). Ancianos: no se han realizado estudios específicos en ancianos. Ahora bien, en los ensayos clínicos realizados con Recormon se incluyó un porcentaje importante de pacientes ancianos, sin que fueran necesarios ajustes especiales de la dosis en este grupo de edad.
Contraindicaciones.
Recormon está contraindicado en pacientes con: hipersensibilidad al principio activo o a cualquiera de los excipientes; hipertensión mal controlada. En la indicación "aumentar el rendimiento de la sangre autóloga", Recormon no debe utilizarse en pacientes que hayan sufrido un infarto de miocardio o un accidente cerebrovascular en el mes anterior al tratamiento, que presenten angina de pecho inestable o que corran el riesgo de trombosis venosa profunda, por ejemplo los que tengan antecedentes de enfermedad tromboembólica venosa. Multidosis/cartuchos solamente: el disolvente contiene alcohol bencílico como conservante y, por tanto, no debe administrarse a lactantes o niños menores de 3 años.
Reacciones adversas.
Ensayos clínicos: de acuerdo con los resultados de los ensayos clínicos en 1.725 pacientes, cabe prever reacciones adversas en aproximadamente un 8% de los pacientes tratados con Recormon. Pacientes con anemia por insuficiencia renal crónica: las reacciones adversas más frecuentes (frecuencia del 1-10%), en especial durante la fase inicial del tratamiento con Recormon, son los episodios hipertensivos consistentes en hipertensión o crisis hipertensivas con o sin síntomas de tipo encefalopático (por ejemplo: cefalea y estado confusional, trastornos sensitivomotores -como alteraciones del habla o deterioro de la marcha- e incluso crisis tonicoclónicas). Estas elevaciones de la tensión arterial pueden producirse en pacientes normotensos o pueden consistir en un agravamiento de una hipertensión ya existente (ver Advertencias). Pueden producirse trombosis de la fístula arteriovenosa, sobre todo en pacientes con tendencia a la hipotensión y en los que hayan presentado complicaciones de la fístula (por ejemplo: estenosis, aneurismas) (ver Advertencias). En la mayoría de los casos, se observa una reducción de los valores séricos de ferritina al mismo tiempo que un aumento de la Hb. También se han descrito casos aislados de aumentos transitorios de potasio y fosfatos en suero. La tabla siguiente recoge la incidencia de las reacciones adversas observada en los ensayos clínicos. Se indica la diferencia existente en la frecuencia de acontecimientos adversos entre los pacientes tratados con Recormon y los del grupo de control.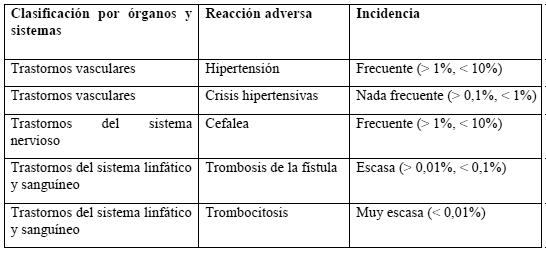
Pacientes con cáncer tratados con quimioterapia que presentan anemia sintomática: los episodios hipertensivos son reacciones adversas frecuentes (1-10%), en especial durante la fase inicial del tratamiento. En algunos pacientes se observa una reducción de los valores de hierro sérico. Los estudios clínicos han mostrado una mayor frecuencia de episodios tromboembólicos en los pacientes con cáncer tratados con Recormon que en los sujetos de control no tratados o tratados con placebo. En los pacientes tratados con Recormon, esta incidencia era del 7%, frente al 4% en los sujetos de control ("frecuente" en ambos casos); ahora bien, no se ha asociado a un aumento de la mortalidad tromboembólica en comparación con los sujetos de control. En la tabla siguiente se presenta la incidencia de las reacciones adversas observada en los ensayos clínicos. Se indica la diferencia existente en la frecuencia de acontecimientos adversos entre los pacientes tratados con Recormon y los del grupo de control.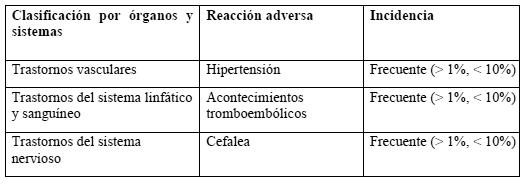
Pacientes que siguen un programa de predonación de sangre autóloga: en estos pacientes se ha descrito una frecuencia ligeramente superior de episodios tromboembólicos, pero no se pudo establecer una relación causal con el tratamiento con Recormon. Puede producirse una ferropenia transitoria (ver Advertencias). En la tabla siguiente se presenta la incidencia de las reacciones adversas observada en los ensayos clínicos. Se indica la diferencia existente en la frecuencia de acontecimientos adversos entre los pacientes tratados con Recormon y los del grupo de control.
Recién nacidos prematuros: la reducción de los valores séricos de ferritina es muy frecuente ( > 10%) (ver Advertencias). Todas las indicaciones: en escasas ocasiones (≥1/10.000 a ≤1/1.000) pueden producirse reacciones cutáneas como erupciones, prurito, urticaria o reacciones en el lugar de inyección. En muy escasas ocasiones (≤1/10.000) se han descrito reacciones anafilactoides. Sin embargo, en estudios clínicos controlados no se observó ningún aumento de la incidencia de hipersensibilidad. En muy escasas ocasiones (≤1/10.000), sobre todo al inicio del tratamiento, se han descrito síntomas de tipo gripal, como fiebre, escalofríos, cefalea, dolor en extremidades, malestar y/o dolor óseo. Estas reacciones fueron de carácter leve o moderado y remitieron tras un par de horas o días. Alteraciones analíticas: ver Advertencias y Pruebas de laboratorio. Experiencia tras la comercialización: se han descrito casos aislados de aplasia eritrocitaria pura (AEP) por anticuerpos neutralizantes antieritropoyetina asociados al tratamiento con Recormon (ver Advertencias). Con la excepción de la aplasia eritrocitaria pura (AEP) por anticuerpos antieritropoyetina, la información sobre seguridad obtenida en la farmacovigilancia tras la comercialización refleja el perfil de acontecimientos adversos esperado en estas poblaciones y el perfil de reacciones adversas de la epoetina beta (ver Advertencias, Uso en poblaciones especiales y Reacciones adversas). Alteraciones analíticas: las anomalías de laboratorio registradas en la farmacovigilancia tras la comercialización reflejan la experiencia obtenida en los ensayos clínicos (ver Advertencias y Ensayos clínicos).
Advertencias.
Recormon debe utilizarse con precaución en presencia de anemia refractaria con exceso de blastos en transformación, epilepsia, trombocitosis o insuficiencia hepática crónica. De igual modo, debe comprobarse que el paciente no sufre una carencia de ácido fólico o vitamina B12, ya que reduciría la eficacia de Recormon. Con objeto de asegurar una eritropoyesis eficaz, debe evaluarse el estado del hierro en todos los pacientes, antes del tratamiento y durante éste, pudiendo ser necesario un tratamiento con suplementos de hierro, que deberá realizarse siguiendo las directrices terapéuticas. Recormon contiene fenilalanina como excipiente. Esto debe tenerse en cuenta en los pacientes con formas graves de fenilcetonuria. Ausencia de efecto: las razones más frecuentes de respuesta incompleta a los agentes estimulantes de la eritropoyesis (AEE) son la ferropenia y la inflamación crónica (por ejemplo, la debida a uremia o cáncer metastásico avanzado). Los siguientes trastornos pueden comprometer también la eficacia del tratamiento con AEE: pérdida hemática crónica, fibrosis de médula ósea, sobrecarga grave de aluminio debida al tratamiento de la insuficiencia renal, carencia de ácido fólico o vitamina B12 y hemólisis. Si se han descartado todos los trastornos citados y el paciente presenta una disminución súbita de la hemoglobina asociada a reticulopenia y anticuerpos antieritropoyetina, debe contemplarse un examen de la médula ósea para el diagnóstico de una posible aplasia eritrocitaria pura (AEP). Si se diagnostica una AEP, debe suspenderse el tratamiento con epoetina y debe pasarse a otro AEE. Se ha descrito anemia eritrocitaria pura causada por anticuerpos neutralizantes antieritropoyetina en asociación con el tratamiento con eritropoyetina, incluido Recormon. Se ha observado que estos anticuerpos presentan una reacción cruzada con todas las proteínas eritropoyéticas, por lo que en los pacientes con sospecha o presencia confirmada de anticuerpos neutralizantes contra la eritropoyetina no debe cambiarse el tratamiento a Recormon (ver Reacciones adversas). Efectos sobre el crecimiento tumoral: las epoetinas son factores de crecimiento que estimulan fundamentalmente la producción de eritrocitos. Los receptores de la eritropoyetina pueden expresarse sobre la superficie de diversas células tumorales. Como con todos los factores de crecimiento, existe la preocupación de que las epoetinas puedan estimular el crecimiento de cualquier tipo de neoplasia maligna. Un estudio clínico controlado, en el que se administró epoetina beta a pacientes con cáncer de cabeza y cuello, ha puesto de manifiesto una supervivencia sin progresión locorregional inferior en pacientes tratados con epoetina beta. Otro estudio clínico en el cáncer de mama, diseñado para identificar un posible efecto positivo de la epoetina beta sobre la supervivencia global en comparación con los controles no tratados, no mostró efectos estadísticamente significativos en la supervivencia global ni en la progresión tumoral. Además, un metanálisis de los datos de estudios clínicos controlados y aleatorizados con epoetina beta en el tratamiento de la anemia en pacientes con cáncer (12 estudios, 2.301 pacientes, incluidos los dos estudios antes mencionados) no reveló ningún efecto negativo estadísticamente significativo sobre la supervivencia o la progresión tumoral (ver Estudios clínicos/eficacia). En pacientes con IRC y pacientes con cáncer tratados con quimioterapia puede producirse un aumento de la tensión arterial (episodios hipertensivos) o un agravamiento de la hipertensión existente, sobre todo en casos de aumento rápido de la Hb. El aumento de la tensión arterial puede tratarse con fármacos antihipertensivos. Si las elevaciones tensionales no pueden controlarse con tratamiento farmacológico, se recomienda una interrupción transitoria del tratamiento con Recormon. En especial al inicio del tratamiento, se recomienda vigilar regularmente la tensión arterial, incluidas las determinaciones entre las sesiones de diálisis en los pacientes con anemia renal. Entre los pacientes con IRC pueden producirse también crisis hipertensivas con síntomas de tipo encefalopático en pacientes que, por lo demás, tienen una tensión arterial normal o baja. Esto requiere la atención inmediata de un médico y cuidados intensivos. Debe prestarse especial atención a las cefaleas lacerantes súbitas de tipo migrañoso como posible señal de alarma al respecto. Una fuerte sobrecarga de aluminio debida al tratamiento de la insuficiencia renal puede menoscabar la eficacia de Recormon. En los pacientes con IRC, a menudo es necesario aumentar la dosis de heparina durante la hemodiálisis mientras dura el tratamiento con Recormon como consecuencia del aumento de la Hb. Puede producirse una oclusión del sistema de diálisis si la heparinización no es óptima. En los pacientes con IRC con riesgo de trombosis de la fístula debe considerarse la revisión temprana de ésta y la profilaxis, por ejemplo con la administración de ácido acetilsalicílico. En los pacientes con IRC puede producirse un aumento moderado y dependiente de la dosis en el recuento plaquetario, dentro de los límites normales, durante el tratamiento con Recormon, en especial tras la administración intravenosa. Esta elevación revierte en el curso del tratamiento. Se recomienda una vigilancia regular del recuento plaquetario durante las 8 primeras semanas de tratamiento. En los pacientes que siguen un programa de predonación de sangre autóloga puede producirse un aumento del recuento plaquetario, generalmente dentro de los límites normales. Por consiguiente, se recomienda determinar el recuento plaquetario al menos una vez por semana en tales pacientes. Si se produce una elevación de las plaquetas de más de 150 x 109/l o si se alcanza un recuento plaquetario superior a los límites normales, debe interrumpirse el tratamiento con Recormon. Cuando se utilice Recormon en un programa de predonación de sangre autóloga, se tendrán en cuenta las directrices oficiales sobre donaciones sanguíneas, particularmente las siguientes: sólo deben donar sangre los pacientes con un hematócrito ≥33% (hemoglobina ≥11g/dl [6,83mmol/l]); - debe procederse con especial cautela si el paciente pesa menos de 50kg; - el volumen de sangre de una extracción no debe superar el 12% del volumen sanguíneo estimado del paciente. El tratamiento debe reservarse para los pacientes en los que se considere de especial importancia evitar transfusiones de sangre homóloga, una vez evaluada la relación riesgos/beneficios de dichas transfusiones. En los pacientes tratados por una anemia de la prematuridad, puede producirse un ligero aumento del recuento plaquetario, en especial hasta el día 12-14 de vida, por lo que debe efectuarse una vigilancia regular de las plaquetas. Abuso farmacológico y dependencia: un mal uso por personas no anémicas puede conducir a un aumento excesivo de la Hb, lo que puede conducir a complicaciones del sistema cardiovascular potencialmente letales. No se han descrito casos de dependencia de la epoetina beta. Efectos sobre la capacidad para conducir y utilizar máquinas: no se han realizado estudios acerca de los efectos sobre la capacidad de conducir y utilizar máquinas. Ahora bien, considerando el mecanismo de acción y el perfil de seguridad conocido de Recormon, no se prevén tales efectos. Pruebas de laboratorio: deben vigilarse regularmente el recuento plaquetario, el hematócrito y la concentración de hemoglobina en todos los pacientes (ver Advertencias). En pacientes con insuficiencia renal crónica se ha descrito una elevación de la concentración sérica de potasio durante el tratamiento con Recormon, aunque no se ha establecido una relación causal. Si se observa una concentración de potasio elevada o en aumento, debe evaluarse la conveniencia de interrumpir la administración de Recormon hasta haberla corregido.
Interacciones.
No se han realizado estudios específicos de interacciones clínicas. La experiencia clínica no muestra ningún indicio de posibles interacciones de Recormon con otros medicamentos (para más información, ver Datos preclínicos sobre seguridad). Uso en poblaciones especiales: embarazo: los estudios realizados con animales no han revelado ningún efecto nocivo directo o indirecto en el embarazo, el desarrollo embrionario/fetal, el parto o el desarrollo postnatal (ver Datos preclínicos sobre seguridad). Toda la información sobre la seguridad de la exposición a Recormon durante el embarazo procede de la experiencia adquirida en la farmacovigilancia tras la comercialización. Un examen de los datos de postcomercialización disponibles no muestra ningún indicio de relación causal entre efectos nocivos para el embarazo, desarrollo embrionario/fetal o desarrollo postnatal y el tratamiento con Recormon. Sin embargo, dada la ausencia de datos de estudios clínicos, es preciso actuar con precaución cuando se prescriba Recormon a mujeres embarazadas. Parto: los estudios realizados con animales no han revelado ningún efecto nocivo directo o indirecto en el embarazo, el desarrollo embrionario/fetal, el parto o el desarrollo postnatal (ver Datos preclínicos sobre seguridad). Toda la información sobre la seguridad de la exposición a Recormon durante el embarazo procede de la experiencia adquirida en la farmacovigilancia tras la comercialización. No se ha observado ningún indicio de efectos nocivos en el parto. Sin embargo, dada la ausencia de datos de estudios clínicos, es preciso actuar con precaución cuando se prescriba Recormon a mujeres embarazadas durante el parto. Lactancia: la experiencia basada en la lactancia humana es tan sólo limitada. La eritropoyetina endógena se excreta en la leche materna y es absorbida con facilidad en el aparato digestivo del recién nacido. La decisión de continuar o interrumpir la lactancia materna o continuar o no el tratamiento con epoetina beta debe tomarse teniendo en cuenta el beneficio para el niño de la lactancia materna y el beneficio para la madre del tratamiento con epoetina beta. Uso en pediatría: se han realizado ensayos clínicos de registro farmacéutico en niños y adolescentes con anemia por insuficiencia renal crónica y en recién nacidos para la prevención de la anemia debida a la prematuridad. En la indicación de anemia por insuficiencia renal crónica, Recormon no debe administrarse a lactantes (es decir, antes de los 2 años de edad) (ver Instrucciones posológicas especiales y Advertencias). En las indicaciones de anemia en pacientes con cáncer tratados con quimioterapia y de tratamiento para aumentar la cantidad de sangre autóloga, Recormon no está indicado en la población pediátrica. Uso en geriatría: ver Instrucciones especiales de posología. Insuficiencia renal: ver Instrucciones especiales de posología. Insuficiencia hepática: no se han realizado ensayos clínicos específicos en pacientes con insuficiencia hepática.
Conservación.
Consérvese en un refrigerador (2-8°C). Viales, cartuchos y jeringas precargadas deben mantenerse en el envase externo para protegerlos de la luz. Liofilizado y disolvente para solución inyectable: con fines de uso ambulatorio, el paciente puede mantener el producto sin reconstituir fuera del refrigerador durante un período único de hasta 5 días a temperatura ambiente (no superior a 25°C). La solución reconstituida debe permanecer fuera del refrigerador solamente el tiempo necesario para preparar las inyecciones. Se ha demostrado la estabilidad química y física de la solución reconstituida durante un mes a 2-8°C. Desde un punto de vista microbiológico, una vez abierta, la solución reconstituida puede mantenerse durante un máximo de un mes a 2-8°C. El usuario será responsable si el tiempo de conservación tras la apertura o las condiciones de conservación son diferentes. Polvo y disolvente para solución inyectable en cartuchos: con fines de uso ambulatorio, el paciente puede mantener fuera del refrigerador el cartucho aún no introducido en el Reco-Pen durante un período único de hasta 5 días, a temperatura ambiente (no superior a 25°C). Se ha demostrado la estabilidad química y física de la solución reconstituida durante un mes a 2-8°C. Desde un punto de vista microbiológico, una vez abierta, la solución reconstituida puede conservarse durante un máximo de un mes a 2-8°C. El usuario será responsable si el tiempo de conservación tras la apertura o las condiciones de conservación son diferentes. Tras la inserción del cartucho en el Reco-Pen, la cadena de frío sólo puede interrumpirse para la administración del producto. Solución inyectable en jeringas precargadas: con fines de uso ambulatorio, el paciente puede mantener el producto sin reconstituir fuera del refrigerador durante un período único de hasta 3 días a temperatura ambiente (no superior a 25°C). Instrucciones especiales de uso, manipulación y eliminación: liofilizado y disolvente para solución inyectable: incompatibilidades: este medicamento no debe diluirse ni mezclarse con otros medicamentos, excepto los mencionados a continuación (contenido de la ampolla de disolvente acompañante). Instrucciones de uso,