RIMENAD
SYNTHON
Inmunomodulador antineoplásico.
Composición.
Cada cápsula contiene: Pomalidomida 4,00 mg. Excipientes c.s.: Celulosa microcristalina, maltodextrina, estearil fumarato de sodio, gelatina, dióxido de titanio, óxido de hierro amarrillo, óxido de hierro rojo, colorante FD&C azul N°2, colorante FD&C rojo N°3, goma laca.
Indicaciones.
En combinación con dexametasona, para pacientes con mieloma múltiple, quienes han recibido al menos dos tratamientos previos que incluyeron lenalidomida y un inhibidor de proteosoma y han demostrado progresión de la enfermedad en o dentro de 60 días de terminado el último tratamiento.
Dosificación.
El tratamiento debe iniciarse y monitorizarse bajo la supervisión de médicos con experiencia en el tratamiento del mieloma múltiple. Posología: La dosis inicial recomendada es de 4 mg de Rimenad una vez al día por vía oral, en los días del 1 al 21 de ciclos repetidos de 28 días. La dosis recomendada de dexametasona es de 40 mg por vía oral una vez al día, en los días 1, 8, 15 y 22 de cada ciclo de tratamiento de 28 días. La posología se mantiene o modifica en función de los resultados clínicos y de laboratorio. El tratamiento debe suspenderse si existe evidencia de progresión de la enfermedad.
Modificación o interrupción de la dosis de pomalidomida. Las instrucciones para la interrupción y reducción de la dosis de pomalidomida relacionadas con reacciones adversas hematológicas se indican en la siguiente tabla: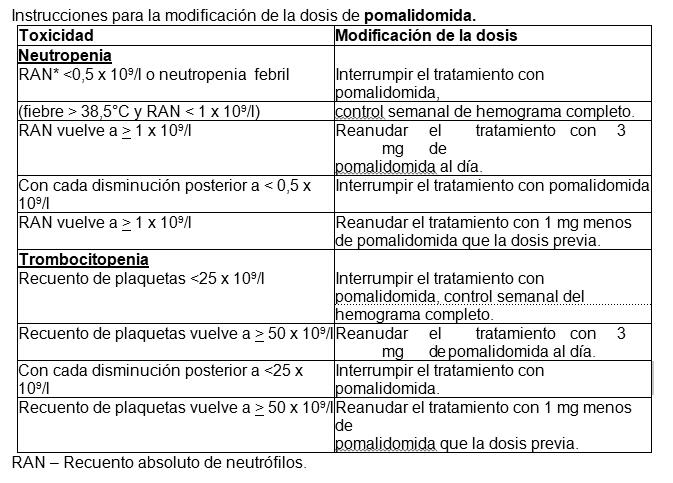
Para iniciar un nuevo ciclo de pomalidomida, el recuento de neutrófilos debe ser ≥1 x 109 /l y el recuento de plaquetas debe ser ≥50 x 109/l. En caso de neutropenia, el médico debe considerar el uso de factores de crecimiento. En el caso de otras reacciones adversas de grado 3 o 4 relacionadas con pomalidomida, el médico debe considerar interrumpir el tratamiento y reanudarlo con un 1 mg menos que la dosis previa una vez que se haya disminuido la reacción adversa a un grado inferior o igual a 2. Si la reacción adversa ocurre tras disminuciones de la dosis a 1 mg, entonces debe suspenderse el tratamiento con este medicamento. Se debe considerar la interrupción o suspensión de pomalidomida en caso de exantema de grado 2-3. Se debe suspender el tratamiento con pomalidomida en caso de angioedema, exantema de grado 4 y exantema ampolloso o exfoliativo, y no se debe reanudar una vez suspendido por estas reacciones. Si se administran inhibidores potentes del CYP1A2 (p. ej., ciprofloxacino, enoxacino y fluvoxamina) de forma concomitante con pomalidomida, se debe reducir la dosis de pomalidomida en un 50%.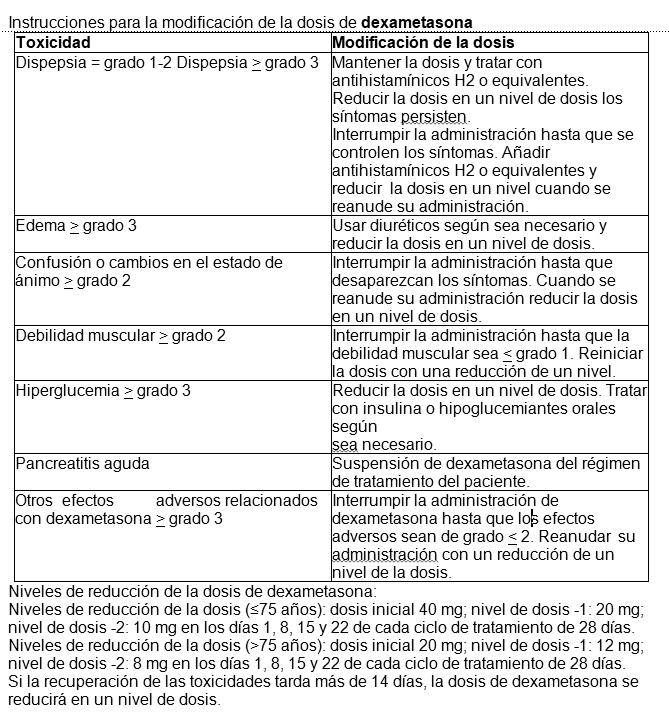
Poblaciones especiales: Población pediátrica: No hay un uso relevante de Rimenad en niños de 0 a 17 años para la indicación del mieloma múltiple. Pacientes de edad avanzada: No se requiere ningún ajuste de dosis de pomalidomida. En pacientes de > 75 años, la dosis inicial de dexametasona es de 20 mg una vez al día en los días 1, 8, 15 y 22 de cada ciclo de tratamiento de 28 días. Insuficiencia renal: No se requiere ningún ajuste de dosis de pomalidomida en los pacientes con insuficiencia renal. Los días de hemodiálisis, los pacientes deben tomar la dosis de pomalidomida después de la hemodiálisis. Insuficiencia hepática: Los pacientes con una concentración sérica de bilirrubina total > 2,0 mg/dl se excluyeron de los estudios clínicos. La insuficiencia hepática tiene un efecto modesto sobre la farmacocinética de pomalidomida. No se requiere ajustar la dosis inicial de pomalidomida en pacientes con insuficiencia hepática según definen los criterios de Child-Pugh. Sin embargo, los pacientes con insuficiencia hepática deben ser monitorizados cuidadosamente por si presentan reacciones adversas y se debe reducir la dosis o suspender la administración de pomalidomida según sea necesario. Forma de administración: Vía oral. Rimenad debe tomarse a la misma hora cada día. Las cápsulas no deben abrirse, romperse ni masticarse. Este medicamento debe tomarse entero, preferiblemente con agua, con o sin alimentos. Si el paciente olvida tomar una dosis de Rimenad un día, el paciente debe entonces tomar la próxima dosis al día siguiente a la hora habitual. Los pacientes no deben ajustar la dosis para compensar una dosis olvidada en días anteriores. Se recomienda presionar solo en un extremo de la cápsula para sacarla del blíster y reducir así el riesgo de deformación o rotura de la cápsula.
Contraindicaciones.
Embarazo. Mujeres con capacidad de gestación, a menos que se consideren medidas de prevención de embarazo. Pacientes varones incapaces de seguir o cumplir las medidas anticonceptivas requeridas. Hipersensibilidad al principio activo o a alguno de los excipientes.
Reacciones adversas.
Al igual que todos los medicamentos, Rimenad puede producir efectos adversos, aunque no todas las personas los sufran. Las reacciones adversas notificadas con mayor frecuencia en los estudios clínicos han sido los trastornos de la sangre y del sistema linfático, incluyendo anemia (45,7%), neutropenia (45,3%) y trombocitopenia (27 %); trastornos generales y alteraciones en el lugar de la administración, incluyendo fatiga (28,3%), pirexia (21%) y edema periférico (13%); e infecciones e infestaciones incluyendo neumonía (10,7%). Las reacciones adversas relacionadas con neuropatía periférica fueron notificadas en el 12,3% de los pacientes y las reacciones adversas de embolismo o tromboembolismo venoso fueron notificadas en el 3,3% de los pacientes. Las reacciones adversas de grado 3 o 4 más frecuentes estaban relacionadas con trastornos de la sangre y del sistema linfático, incluyendo neutropenia (41,7%), anemia (27%) y trombocitopenia (20,7%); infecciones e infestaciones, incluyendo neumonía (9%); y trastornos generales y alteraciones en el lugar de la administración, incluyendo fatiga (4,7%), pirexia (3%) y edema periférico (1,3%). La reacción adversa grave notificada con mayor frecuencia fue la neumonía (9,3%). Otras reacciones adversas graves notificadas incluyen neutropenia febril (4,0%), neutropenia (2,0%), trombocitopenia (1,7%) y reacciones adversas de TEV (1,7%). Se observó que las reacciones adversas tendían a ocurrir con mayor frecuencia dentro de los primeros 2 ciclos de tratamiento con pomalidomida. Lista tabulada de reacciones adversas: En el estudio aleatorizado CC-4047-MM-003, un total de 302 pacientes con mieloma múltiple en recaída y refractario fueron tratados con 4 mg de pomalidomida administrada una vez al día durante 21 días en cada ciclo de 28 días, en combinación con una dosis baja semanal de dexametasona. Las reacciones adversas observadas en los pacientes tratados con pomalidomida y dexametasona se incluyen a continuación, según el sistema de clasificación por órganos y la frecuencia (SOC por sus siglas en inglés) para todas las reacciones adversas y para las reacciones adversas de Grado 3 o 4. Las frecuencias de las reacciones adversas son las notificadas en el grupo de pomalidomida más dexametasona del estudio CC-4047-MM-003 (n=302). Las reacciones adversas se presentan en orden decreciente de gravedad dentro de cada intervalo de SOC (por sus siglas en inglés) y de frecuencia. Las frecuencias se definen, según las guías actuales, como: muy frecuentes (≥1/10); frecuentes (≥1/100 a < 1/10) y poco frecuentes (≥1/1.000 a < 1/100).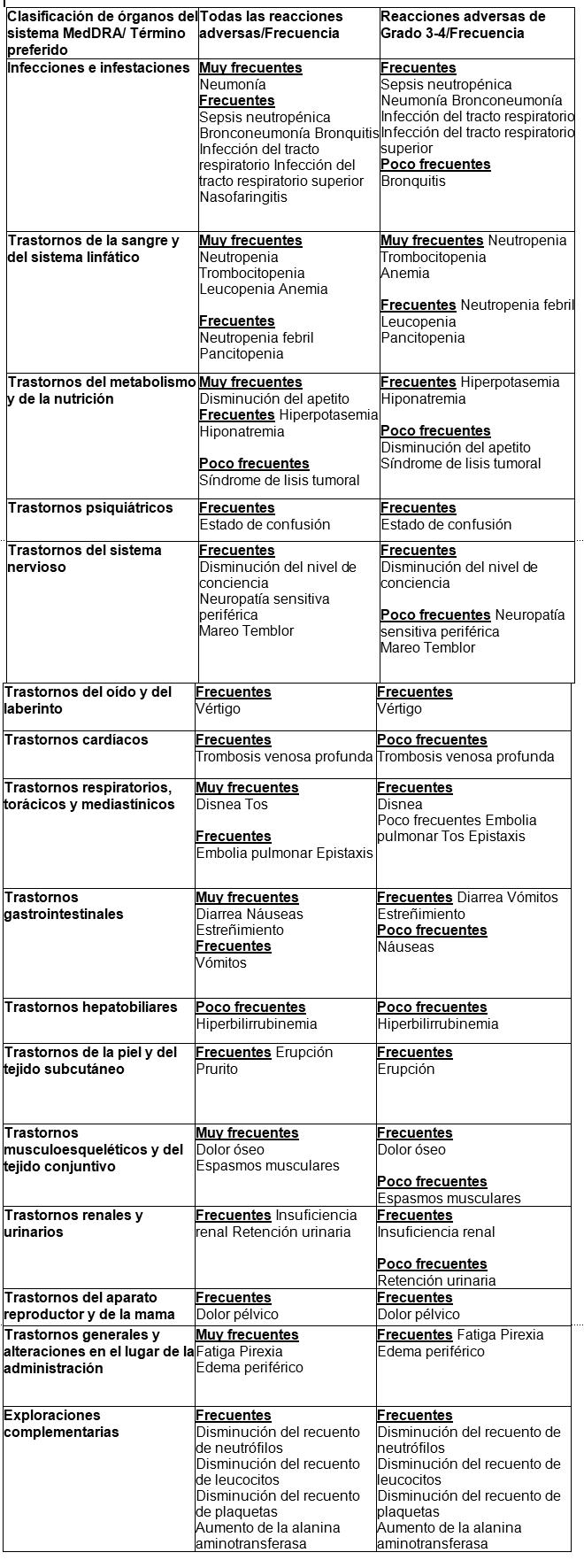
Teratogenicidad: Pomalidomida está relacionada estructuralmente con talidomida. Talidomida es un principio activo con acción teratogénica conocida en humanos, que causa defectos de nacimiento graves que pueden poner en peligro la vida del niño. Pomalidomida tiene un efecto teratogénico en ratas y conejos cuando se administra durante el periodo de mayor organogénesis. Si se toma pomalidomida durante el embarazo, se espera un efecto teratogénico de pomalidomida en los seres humanos.
Presentación.
Est Cap x 21
Nota.
Bioequivalente: Equivalente terapéutico.