TARCEVA®
ROCHE
Agente antineoplásico.
Composición.
Principio activo: clorhidrato de erlotinib. Cada comprimido recubierto de cada dosis contiene clorhidrato de erlotinib en una cantidad equivalente a 25 mg, 100 mg o 150 mg de erlotinib. Lista de excipientes: Núcleo de los comprimidos: Lactosa, monohidrato Ph. Eur./USP/JP. Celulosa microcristalina Ph. Eur./USP/JP. Glicolato de sodio y almidón Ph. Eur./USP/JPE. Laurilsulfato de sodio Ph. Eur./USP/JP. Estearato de magnesio Ph. Eur./USP/JP. Recubrimiento de los comprimidos: Hidroxipropilcelulosa Ph. Eur./USP/JP. Dióxido de titanio Ph. Eur./USP/JP. Polietilenglicol Ph. Eur./NF/JP. Hipromelosa Ph. Eur./USP/JP. Forma farmacéutica: Tarceva en comprimidos recubiertos de 25 mg, 100 mg y 150 mg. Comprimidos recubiertos de color blanco o amarillento, redondos, biconvexos. El color de la impresión varía según la dosis de los comprimidos. Vía de administración: Oral (p.o.). Declaración de esterilidad / radiactividad: No procede.
Farmacología.
Propiedades farmacodinámicas: Mecanismo de acción: El erlotinib es un potente inhibidor de la fosforilación intracelular del receptor HER1/EGFR. El receptor HER1/EGFR se expresa en la superficie celular de células normales y células cancerosas. En modelos no clínicos, la inhibición de la fosforilación del EGFR se traduce en estasis y/o muerte celular. Ensayos clínicos / Eficacia: Carcinoma pulmonar no microcítico (Tarceva en monoterapia): Tratamiento de primera línea de pacientes con mutaciones activadoras de EGFR: La eficacia de Tarceva como tratamiento de primera línea de pacientes con CPNM y mutaciones activadoras de EGFR ha quedado demostrada en un estudio de fase III abierto y aleatorizado (ML20650, EURTAC). Este estudio se realizó en pacientes de raza blanca con CPNM metastásico o localmente avanzado (estadios IIIB y IV) que no habían recibido previamente quimioterapia o algún tratamiento antineoplásico sistémico contra su enfermedad avanzada y que presentaban mutaciones en el dominio tirosincinasa del gen EGFR (deleción del exón 19 o mutación del exón 21). Se aleatorizó a los pacientes en la relación 1:1 para recibir 150 mg de Tarceva o biquimioterapia basada en el platino. La variable principal de valoración, la supervivencia sin progresión (SSP) evaluada por el investigador, se determinó mediante un análisis intermedio preestablecido (n = 153, hazard ratio [HR]: 0,42; IC del 95 %: 0,27-0,64; p < 0,0001 en el grupo de Tarceva [n = 77] en relación con el grupo de quimioterapia [n = 76]). Se observó una reducción del 58% del riesgo de progresión de la enfermedad o de fallecimiento. La mediana de SSP fue de 9,4 meses en el grupo de Tarceva y de 5,2 meses en el de quimioterapia; y la tasa de respuesta objetiva, del 54,5 % y el 10,5%, respectivamente. Los resultados de la SSP fueron confirmados por un Comité independiente que evaluó los estudios tomográficos: la mediana de SSP fue de 10,4 meses en el grupo de Tarceva frente a 5,4 meses en el de quimioterapia (HR: 0,47; IC del 95 %: 0,27-0,78; p = 0,003). Los datos de la supervivencia global no estaban maduros en el momento del análisis intermedio (HR: 0,80; IC del 95%: 0,47-1,37, p = 0,4170). Otros datos publicados: En un análisis prospectivo de pacientes con CPNM avanzado cuyos tumores presentaban mutaciones activadoras en el dominio TK de EGFR, la mediana de SSP en los 113 pacientes tratados con Tarceva en primera línea fue de 14 meses (IC del 95%: 9,7-18,3 meses), y la mediana de supervivencia global, de 28,0 meses (IC del 95%: 22,7-33 meses). Un análisis agrupado de los datos publicados de pacientes con CPNM reveló que en los pacientes que presentaban tumores con mutaciones activadoras de EGFR y habían recibido Tarceva como tratamiento predominante de primera línea (n = 70, 12,5 meses, IC del 95%: 10,6-16,0) era más larga la mediana de SSP que en los que habían recibido quimioterapia (n = 359, 6,0 meses, IC del 95%: 5,4-6,7). Terapia de mantenimiento de primera línea: La eficacia y la seguridad de Tarceva como terapia de mantenimiento de primera línea en el CPNM quedó demostrada en un estudio clínico aleatorizado, de doble-ciego y controlado con placebo (BO18192). Este estudio se realizó en 889 pacientes con CPNM localmente avanzado o metastásico que no había progresado durante 4 ciclos de biquimioterapia basada en el platino. Se aleatorizó a los pacientes en la relación 1:1 para recibir 150 mg de Tarceva o placebo por vía oral, una vez al día. La variable principal de valoración del estudio era la SSP en todos los pacientes y en los pacientes con tumor positivo a la prueba de expresión de EGFR por IHC (inmunohistoquímica). Las características basales demográficas y patológicas estaban bien equilibradas entre los dos grupos de tratamiento. La Tabla 3 recoge los resultados del análisis principal de la SSP en la población con intención de tratar (ITT). El análisis principal de la SSP en todos los pacientes reveló una hazard ratio para la SSP en el grupo de Tarceva de 0,71 en relación con el grupo de placebo. La SSP media fue de 22,4 semanas en el grupo de Tarceva y de 16,0 semanas en el de placebo.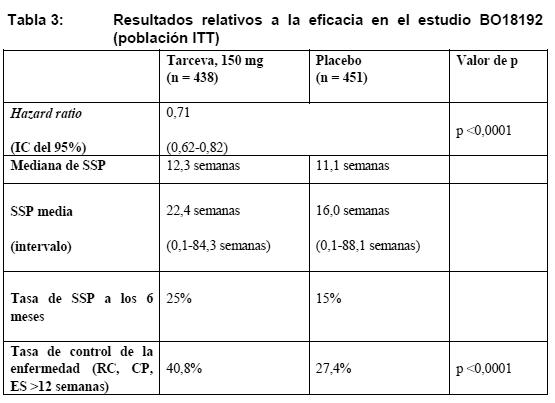
El análisis principal de la SSP en los pacientes con tumor positivo a la prueba de expresión de EGFR por IHC fue similar, con una hazard ratio de 0,69 (IC del 95%: 0,58-0,82) (p < 0,0001). La SSP media fue de 22,8 semanas en el grupo de Tarceva (intervalo: 0,1-78,9 semanas) y de 16,2 semanas en el de placebo (intervalo: 0,1-88,1 semanas). El porcentaje de pacientes sin progresión a 6 meses fue del 27% y el 16%, respectivamente. La eficacia, basada en la estratificación y los factores clínicos, fue homogénea en todos los subgrupos. Beneficio terapéutico se observó en todos los subgrupos de biomarcadores, independientemente de la expresión de EGFR determinada por IHC y por FISH, la mutación de EGFR o el estado mutacional de K-ras. Aunque la activación de mutaciones de EGFR identifica a los pacientes con el beneficio más grande (HR: 0,10, p < 0,0001), estas mutaciones no son requisito para un efecto, puesto que también se ha observado beneficio en pacientes con EGFR de tipo natural (wild) (HR: 0,78, p = 0,0185). La calidad de vida no apuntaba a ningún efecto pernicioso del erlotinib en comparación con placebo. El beneficio en la SSP se tradujo en un beneficio estadísticamente significativo y clínicamente importante en la supervivencia global (variable secundaria de valoración, HR: 0,81; v. Tabla 4).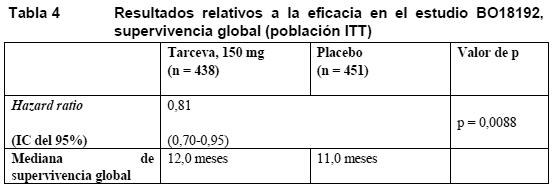
Tratamiento de segunda/tercera línea: La eficacia y la seguridad de Tarceva como tratamiento de segunda/tercera línea del CPNM quedó demostrada en un estudio clínico aleatorizado, de doble-ciego y controlado con placebo (BR.21). Este estudio se realizó en 17 países y en él participaron 731 pacientes con CPNM localmente avanzado o metastásico tras el fracaso de al menos una pauta de quimioterapia. Se aleatorizó a los pacientes en la relación 2:1 para recibir 150 mg de Tarceva o placebo por vía oral, una vez al día. Las variables de valoración del estudio eran: supervivencia global, tiempo hasta el deterioro de síntomas relacionados con el cáncer pulmonar (tos, disnea, dolor), tasa de respuesta, duración de la respuesta, SSP y seguridad. La variable principal de valoración era la supervivencia. Dada la aleatorización 2:1, 488 pacientes se asignaron al grupo de Tarceva y 243 al de placebo. No se seleccionó a los pacientes por el estado de HER1/EGFR, el sexo, la raza, los antecedentes de tabaquismo ni la clasificación histológica. Las características demográficas estaban bien equilibradas entre los dos grupos de tratamiento. Aproximadamente dos tercios de los pacientes eran de sexo masculino y aproximadamente un tercio presentaban un estado general basal según la escala ECOG de 2, y el 9%, de 3. El 93% y el 92% de todos los pacientes de los grupos de Tarceva y placebo, respectivamente, habían recibido anteriormente al menos una pauta de quimioterapia con platino, y el 36% y el 37%, respectivamente, habían recibido anteriormente tratamiento con taxanos. El 50% de los pacientes habían recibido anteriormente sólo al menos una pauta de quimioterapia. Se evaluó la supervivencia en la población con intención de tratar. La mediana de supervivencia global mejoró en un 42,5% y fue de 6,7 meses en el grupo de Tarceva (IC del 95%: 5,5-7,8 meses), frente a 4,7 meses en el grupo de placebo (IC del 95%: 4,1-6,3 meses). El análisis principal de la supervivencia se ajustó por los factores de estratificación según los datos en el momento de la aleatorización (estado general según la escala ECOG, mejor respuesta antes del tratamiento, número de regímenes anteriores y exposición previa al platino) y el estado de HER1/EGFR. En el análisis principal, la hazard ratio ajustada por fallecimientos en el grupo de Tarceva en comparación con el grupo del placebo fue de 0,73 (IC del 95%: 0,60-0,87) (p = 0,001). El porcentaje de pacientes vivos a 12 meses fue del 31,2% y el 21,5%, respectivamente. La mejora de la supervivencia con Tarceva se observó en la mayoría de los subgrupos. Para evaluar la robustez del resultado de la supervivencia global, en análisis unifactoriales exploratorios se examinó a una serie de subgrupos de pacientes formados por los valores de los factores de estratificación en la aleatorización y en la situación basal, el estado de HER1/EGFR, la exposición previa a taxanos, los antecedentes de tabaquismo, el sexo, la edad, la histología, la pérdida de peso anterior, el tiempo entre el diagnóstico inicial y la aleatorización y la localización geográfica. Casi todas las hazard ratios en el grupo de Tarceva en relación con el grupo de placebo eran inferiores a 1,0, lo cual era indicativo de una mejora robusta de la supervivencia con Tarceva en todos los subgrupos. Es de destacar que la mejora de la supervivencia con Tarceva era comparable en los pacientes con un estado general según la escala ECOG de 2-3 (HR: 0,77) o un estado general de 0-1 (HR: 0,73) y los pacientes que habían recibido una pauta de quimioterapia (HR: 0,76) o dos o más pautas (HR: 0,76). También se observó una mejora de la supervivencia con Tarceva en los pacientes sin una remisión tumoral objetiva (según los criterios RECIST), como evidenciaba una hazard ratio de fallecimiento de 0,83 en los pacientes cuya mejor respuesta era enfermedad estable y de 0,85 en los pacientes cuya mejor respuesta era enfermedad progresiva. La Tabla 5 resume los resultados del estudio, incluida la supervivencia, el tiempo hasta el deterioro de síntomas relacionados con el cáncer pulmonar y la supervivencia sin progresión.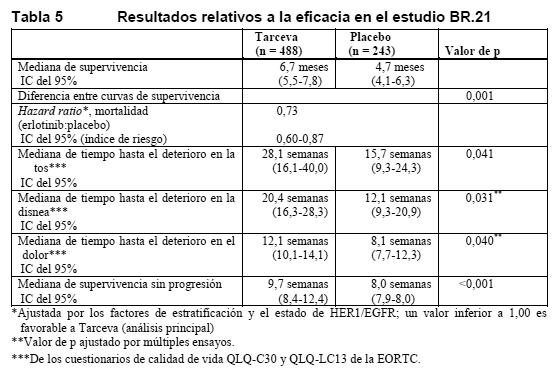
El deterioro sintomatológico se determinó mediante los Cuestionarios de calidad de vida QLQ-C30 y QLQ-LC13 de la EORTC. Los valores basales de tos, disnea y dolor eran similares en los dos grupos de tratamiento. Con Tarceva se obtuvo una mejora de los síntomas consistente en un alargamiento significativo del tiempo hasta el deterioro de la tos (HR: 0,75), la disnea (HR: 0,72) y el dolor (HR: 0,77) en comparación con placebo. Esta mejora sintomatológica no se debió a una mayor utilización de radioterapia paliativa o medicación concomitante en el grupo de Tarceva. La mediana de SSP fue de 9,7 semanas en el grupo de Tarceva (IC del 95%, 8,4-12,4 semanas) frente a 8,0 semanas en el de placebo (IC del 95%, 7,9-8,1 semanas). La HR de la progresión, ajustada por los factores de estratificación y el estado de HER1/EGFR, fue de 0,61 (IC del 95%: 0,51-0,73) (p < 0,001). La tasa de SSP a los 6 meses fue del 24,5% y el 9,3% en los grupos de Tarceva y placebo, respectivamente. La tasa de respuesta objetiva según los criterios RECIST fue del 8,9% en el grupo de Tarceva (IC del 95%: 6,4-12,0%). La mediana de duración de la respuesta fue de 34,3 semanas: entre 9,7 y 57,6+ semanas. En el grupo de placebo se notificaron dos respuestas (0,9%, IC del 95%: 0,1-3,4). La proporción de pacientes con respuesta completa, respuesta parcial o enfermedad estable fue del 44,0% y el 27,5%, en los grupos de Tarceva y placebo, respectivamente (p = 0,004). Carcinoma pancreático (Tarceva administrado simultáneamente con gemcitabina): La eficacia y la seguridad de Tarceva en combinación con gemcitabina como tratamiento de primera línea se evaluó en un estudio aleatorizado, de doble-ciego y controlado con placebo en 569 pacientes con carcinoma pancreático localmente avanzado, irresecable o metastásico. Se aleatorizó 1:1 a los pacientes para recibir Tarceva (100 mg o 150 mg) o placebo una vez al día en una pauta continua junto con gemcitabina i.v. (1.000 mg/m2, ciclo 1: días 1, 8, 15, 22, 29, 36 y 43 de un ciclo de 8 semanas; ciclo 2 y ciclos siguientes: días 1, 8 y 15 de un ciclo de 4 semanas [dosis y pauta aprobadas para el carcinoma pancreático: véase el resumen de características del producto de la gemcitabina]). Tarceva y placebo se tomaron por vía oral, una vez al día, hasta la progresión de la enfermedad o una toxicidad inaceptable. Las variables de valoración del estudio eran la supervivencia global, la tasa de respuesta y la supervivencia sin progresión. También se examinó la duración de la respuesta. La variable principal de valoración era la supervivencia. Se aleatorizó a un total 285 pacientes para recibir gemcitabina + Tarceva (261 pacientes en la cohorte de 100 mg y 24 pacientes en la de 150 mg) y a 284 pacientes para recibir gemcitabina + placebo (260 pacientes en la cohorte de 100 mg y 24 pacientes en la de 150 mg). En la cohorte de 150 mg fueron demasiado pocas las observaciones para extraer conclusiones. Las características basales tanto demográficas como patológicas eran similares en los 2 grupos de tratamiento, 100 mg de Tarceva + gemcitabina o placebo + gemcitabina, salvo en la proporción ligeramente mayor de mujeres en el grupo de Tarceva (51%) que en el de placebo (44%). La mediana de tiempo entre el diagnóstico inicial y la aleatorización fue de aproximadamente 1,0 mes. Aproximadamente la mitad de los pacientes presentaban un estado general basal según la escala ECOG de 1, y el 17%, de 2. La mayoría de los pacientes presentaban metástasis al incorporarse al estudio como manifestación inicial del carcinoma pancreático (77% en el grupo de Tarceva y 76% en el de placebo). La supervivencia se evaluó en la población con intención de tratar a partir de los datos de supervivencia durante el seguimiento, que comprendían 551 muertes. Se presentan los resultados obtenidos en el grupo con la dosis de 100 mg (504 muertes). La hazard ratio ajustada de muerte en el grupo de Tarceva en comparación con el de placebo fue de 0,82 (IC del 95 %: 0,69-0,98) (p = 0,028). El porcentaje de pacientes vivos a los 12 meses fue del 23,8% y el 19,4% en los grupos de Tarceva y placebo, respectivamente. La mediana de supervivencia global fue de 6,4 y 6 meses en los grupos de Tarceva y placebo, respectivamente. En la Tabla 6 se resumen los resultados del estudio.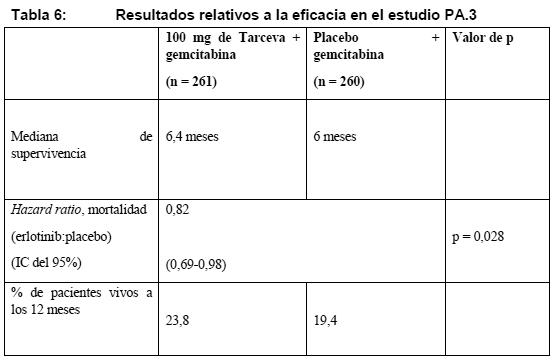
La mediana de SSP fue de 3,81 meses (16,5 semanas) en el grupo de Tarceva (IC del 95 %: 3,58-4,93 meses) frente a 3,55 meses (15,2 semanas) en el grupo de placebo (IC del 95 %: 3,29-3,75 meses) (p = 0,006). La mediana de duración de la respuesta fue de 23,9 semanas: entre 3,71 y 56+ semanas. La tasa de respuesta objetiva (respuesta completa y respuesta parcial) fue del 8,6% y el 7,9% en los grupos de Tarceva y placebo, respectivamente. La proporción de pacientes con respuesta completa, respuesta parcial o enfermedad estable fue del 59% y el 49,4% en los grupos de Tarceva y placebo, respectivamente (p = 0,036). Propiedades farmacocinéticas: Exposición: Tras una dosis oral de 150 mg de Tarceva, la mediana de tiempo hasta la concentración plasmática máxima en equilibrio es de aproximadamente 4,0 horas, y la mediana de concentración plasmática máxima, de 1.995 ng/ml. Antes de la dosis siguiente a las 24 horas, la mediana de concentración plasmática mínima es de 1.238 ng/ml. La mediana de ABC en equilibrio en el intervalo entre dosis es de 41,300 m·h/ml. Absorción: El erlotinib se absorbe bien por vía oral. Su fase de absorción es extensa, alcanzando la concentración plasmática máxima a las 4 horas, por término medio, de la administración oral. La biodisponibilidad estimada en un estudio en voluntarios sanos era del 59%. La exposición tras la administración oral puede aumentar con los alimentos. Tras su absorción, el erlotinib se liga en alto grado en la sangre: aproximadamente el 95% se une a componentes sanguíneos, sobre todo proteínas plasmáticas (albúmina y alfa-1- glucoproteína ácida), con una fracción libre del 5% aproximadamente. Distribución: El erlotinib tiene un volumen de distribución aparente medio de 232 litros y se distribuye en el tejido tumoral humano. En un estudio en 4 pacientes (3 con CPNM y 1 con cáncer de laringe) tratados con dosis orales diarias de 150 mg de Tarceva, la concentración media de erlotinib en muestras de tumores obtenidas quirúrgicamente el día 9 de tratamiento fue de 1.185 ng/g de tejido tumoral. Este valor correspondía a un promedio global del 63 % de las concentraciones plasmáticas máximas en equilibrio. Los metabolitos activos principales estaban presentes en el tumor en concentraciones medias de 160 ng/g de tejido, lo que corresponde a un promedio global del 113 % de las concentraciones plasmáticas máximas en equilibrio. Los estudios de distribución hística utilizando la autorradiografía de cuerpo entero tras la administración oral de erlotinib marcado con [14C] en ratones atímicos con xenoinjertos de tumor HN5 han demostrado una distribución rápida y amplia, con concentraciones máximas de fármaco radiomarcado (aproximadamente el 73% de los valores en plasma) al cabo de 1 hora. Metabolismo: En el ser humano, el erlotinib lo metabolizan citocromos P-450 hepáticos, fundamentalmente CYP3A4 y en menor grado CYP1A2. Es posible que la metabolización extrahepática por el CYP3A4 en el intestino, el CYP1A1 en los pulmones y el CYP1B1 en el tejido tumoral contribuya también al aclaramiento metabólico del erlotinib. Los estudios in vitro indican que aproximadamente el 80-95% del metabolismo del erlotinib corresponde al CYP3A4. Se han identificado tres vías metabólicas principales: 1) O-desmetilación de una cadena lateral o de ambas, seguida de la oxidación de los ácidos carboxílicos; 2) oxidación de la porción acetilénica seguida de la hidrólisis del ácido aril-carboxílico, y 3) hidroxilación aromática de la porción fenilacetilénica. Los metabolitos principales del erlotinib que se produjeron por O-desmetilación de alguna cadena lateral tienen una potencia comparable a la del erlotinib en los ensayos preclínicos in vitro y en los modelos tumorales in vivo. Están presentes en el plasma en concentraciones < 10% de la concentración de erlotinib y tienen una farmacocinética similar a la del erlotinib. Eliminación: Los metabolitos y cantidades traza de erlotinib se excretan predominantemente con las heces ( > 90%), eliminándose por el riñón sólo una pequeña cantidad de la dosis oral. Aclaramiento: Un análisis farmacocinético poblacional en 591 pacientes que recibieron Tarceva en monoterapia reveló un aclaramiento aparente medio de 4,47 l/hora, con una mediana de semivida de 36,2 horas. Por tanto, es previsible que el período hasta la concentración plasmática en equilibrio sea de 7-8 días. No se observaron relaciones significativas entre el aclaramiento aparente previsto y la edad, el peso, el sexo o la raza de los pacientes. Los factores del paciente que se correlacionan con la farmacocinética del erlotinib son la bilirrubina total en suero, las concentraciones de alfa-1-glucoproteína ácida y el tabaquismo vigente. El aumento de las concentraciones séricas de bilirrubina total y las concentraciones de alfa-1-glucoproteína ácida se asociaron a una menor tasa de aclaramiento del erlotinib. Los fumadores presentaban una tasa mayor de aclaramiento del erlotinib (ver Interacciones). Se realizó un segundo análisis de farmacocinética poblacional en el que se incluyeron los datos del erlotinib correspondientes a 204 pacientes con carcinoma pancreático tratados con erlotinib + gemcitabina. En este análisis se demostró que las covariables que afectaban al aclaramiento del erlotinib en los pacientes del estudio del carcinoma pancreático eran muy similares a las observadas en el análisis farmacocinético previo de la monoterapia. No se identificó ningún nuevo efecto de covariables. La coadministración de gemcitabina no tuvo ningún efecto en el aclaramiento plasmático del erlotinib. Farmacocinética en poblaciones especiales: No se han realizado estudios específicos en niños ni en ancianos. Insuficiencia hepática: El aclaramiento del erlotinib es principalmente renal. La exposición al erlotinib era similar en los pacientes con disfunción hepática moderada (índice de Child-Pugh 7-9) y en los que presentaban una función hepática adecuada, incluidos aquellos con carcinoma primario de hígado o metástasis hepáticas. Insuficiencia renal: El erlotinib y sus metabolitos no se excretan en grado significativo por los riñones ( < 9% de una dosis única se excreta con la orina). No se han llevado a cabo estudios clínicos en pacientes con la función renal menoscabada. Fumadores: Un estudio farmacocinético en sujetos sanos no fumadores y fumadores vigentes de cigarrillos ha revelado que fumar cigarrillos incrementa el aclaramiento del erlotinib y disminuye la exposición al mismo. El ABC0-inf en los fumadores era aproximadamente 1/3 del de los nunca fumadores o ex fumadores (n = 16 en cada grupo de los nunca fumadores y ex fumadores). Esta exposición reducida en los fumadores vigentes se debe presumiblemente a la inducción de CYP1A1 en los pulmones y de CYP1A2 en el hígado. En el estudio fundamental de fase III en el CPNM, los fumadores vigentes alcanzaron una concentración plasmática mínima en equilibrio de 0,65 mg/ml (n = 16), la cual era aproximadamente el doble de baja que la de los ex fumadores o los pacientes que nunca habían fumado (1,28 mg/ml, n = 108). Este efecto se acompañó de un aumento del 24% del aclaramiento plasmático aparente del erlotinib. En un estudio de fase I de escalonamiento de la dosis en pacientes con CPNM que eran fumadores vigentes, los análisis farmacocinéticos en estado de equilibrio mostraban un aumento proporcional a la dosis de la exposición al erlotinib cuando la dosis de Tarceva se incrementaba de 150 mg a la dosis máxima tolerada de 300 mg. En este estudio, la concentración plasmática mínima en equilibrio con una dosis de 300 mg en los fumadores vigentes fue de 1,22 mg/ml (n = 17) (ver Pautas posológicas especiales e Interacciones). Datos preclínicos sobre seguridad: Carcinogenicidad: En los estudios preclínicos no se han observado indicios de potencial carcinógeno. El erlotinib no fue ni genotóxico ni clastógeno en los estudios de toxicidad genética. Los resultados de estudios de carcinogenicidad de dos años del erlotinib en ratas y ratones con una exposición superior a la terapéutica para el ser humano fueron negativos. Mutagenicidad: Los resultados del erlotinib en las pruebas estándares de genotoxicidad fueron negativos. Trastornos de la fecundidad: No se observaron alteraciones de la fecundidad en los estudios realizados en ratas macho y hembra con dosis próximas a la máxima tolerada. Teratogenicidad: Los datos de las pruebas de toxicología en la reproducción en ratas y conejos indican que, tras la exposición al erlotinib en dosis próximas a la dosis máxima tolerada y dosis que eran tóxicas para la madre, se produjo embriotoxicidad, pero no hubo signos de alteración de la fertilidad, de teratogénesis ni de anomalías del desarrollo físico o conductual prenatal o posnatal. La toxicidad materna en ratas y conejos en estos estudios se produjo con exposiciones plasmáticas similares a las obtenidas en el ser humano con dosis de 150 mg de erlotinib. Otros efectos: Los efectos de la administración a largo plazo observados al menos en una especie animal o un estudio incluyeron los efectos sobre la córnea (atrofia, ulceración), la piel (degeneración e inflamación folicular, enrojecimiento y alopecia), los ovarios (atrofia), el hígado (necrosis hepática), los riñones (necrosis papilar renal y dilatación tubular) y el tubo digestivo (retraso en el vaciamiento gástrico y diarrea). El recuento de glóbulos rojos, el hematócrito y la hemoglobina disminuyeron, y el recuento de reticulocitos aumentó. El recuento de glóbulos blancos, sobre todo de neutrófilos, creció. Se produjo un aumento relacionado con el tratamiento de la alanina-aminotransferasa (ALAT), la aspartato-aminotransferasa (ASAT) y la bilirrubina. Los estudios in vitro del erlotinib revelaron una inhibición de los canales hERG con concentraciones al menos 20 veces mayores que la concentración de fármaco libre en el ser humano con dosis terapéuticas. Los estudios en perros no evidenciaron una prolongación del segmento QT. En una revisión sistemática centralizada de los datos electrocardiográficos de 152 individuos de siete estudios en voluntarios sanos no se hallaron signos de prolongación del segmento QT, como tampoco se han encontrado en los estudios clínicos signos de arritmias, asociados a la prolongación del segmento QT.
Indicaciones.
Carcinoma pulmonar no microcítico: Tarceva está indicado como tratamiento de primera línea del carcinoma pulmonar no microcítico (CPNM) localmente avanzado o metastásico con mutaciones activadoras de EGFR. Tarceva está indicado como tratamiento de mantenimiento del CPNM localmente avanzado o metastásico que no haya progresado con la quimioterapia de primera línea. Tarceva está indicado asimismo para el tratamiento del CPNM localmente avanzado o metastásico tras el fracaso de al menos una pauta de quimioterapia anterior. Carcinoma pancreático: Tarceva en combinación con gemcitabina está indicado como tratamiento de primera línea del carcinoma pancreático localmente avanzado, irresecable o metastásico.
Dosificación.
Dosis habitual: Carcinoma pulmonar no microcítico: En los pacientes con CPNM avanzado o metastásico sin tratamiento quimioterápico previo debe realizarse la prueba de mutaciones EGFR antes de empezar el tratamiento con Tarceva. La dosis diaria recomendada de Tarceva es de 150 mg, administrada al menos una hora antes o dos horas después de una comida. Carcinoma pancreático: La dosis diaria recomendada de Tarceva es de 100 mg, administrada al menos una hora antes o dos horas después de una comida, en combinación con gemcitabina (ver el resumen de características del producto de la gemcitabina para la indicación carcinoma pancreático). Pautas posológicas especiales: El uso concomitante de sustratos y moduladores del citocromo CYP3A4 puede requerir un ajuste de la dosis (ver Interacciones). Cuando sea necesario ajustar la dosis, se recomienda reducirla en fracciones de 50 mg (ver Advertencias e Interacciones). Insuficiencia hepática: El erlotinib se elimina por metabolización hepática y excreción biliar. Aunque la exposición al erlotinib era similar en los pacientes con disfunción hepática moderada (índice de Child-Pugh 7-9) y los pacientes con una función hepática adecuada, la administración de Tarceva a pacientes con insuficiencia hepática requiere precaución especial. Si se producen reacciones adversas graves, se debe considerar una reducción de la dosis o suspender la administración de Tarceva. La seguridad y la eficacia no se han estudiado en pacientes con insuficiencia hepática grave (ver Advertencias [Hepatitis, insuficiencia hepática] y Farmacocinética en poblaciones especiales). Insuficiencia renal: No se han estudiado la seguridad ni la eficacia de Tarceva en pacientes con insuficiencia renal (ver Farmacocinética en poblaciones especiales). Uso en pediatría: No se han estudiado la seguridad ni la eficacia de Tarceva en pacientes menores de 18 años. Fumadores: Está demostrado que el tabaquismo reduce la exposición al erlotinib en un 50-60%. La dosis máxima tolerada de Tarceva por pacientes con CPNM que eran fumadores vigentes de cigarrillos ha sido de 300 mg. No se conocen la eficacia ni la seguridad a largo plazo de una dosis superior a las dosis iniciales recomendadas en los pacientes que siguen fumando cigarrillos (ver Interacciones y Farmacocinética en poblaciones especiales).
Contraindicaciones.
Tarceva está contraindicado en caso de hipersensibilidad grave al erlotinib o a cualquier otro de sus componentes.
Reacciones adversas.
Ensayos clínicos: La evaluación de la seguridad de Tarceva se basa en los datos de más de 1.200 pacientes tratados con al menos una dosis de 150 mg de Tarceva en monoterapia y los de más de 300 pacientes que recibieron dosis de 100 mg o 150 mg de Tarceva en combinación con gemcitabina. En las tablas siguientes se resume la incidencia de reacciones adversas (RA) notificadas en los estudios clínicos con Tarceva en monoterapia o en combinación con quimioterapia. Se enumeran las RA notificadas en al menos el 10% (grupo de Tarceva) de los pacientes y con una frecuencia superior (≥ 3%) en los tratados con Tarceva que en el grupo de comparación. Tarceva en monoterapia: Las RA de la Tabla 1 se basan en los datos de un estudio de doble-ciego aleatorizado (BR.21) en 731 pacientes con CPNM localmente avanzado o metastásico tras el fracaso de al menos una pauta de quimioterapia. Se aleatorizó a los pacientes en la relación 2:1 para recibir 150 mg de Tarceva o placebo. El medicamento en estudio se administró p.o., una vez al día, hasta la progresión de la enfermedad o una toxicidad inaceptable. Las RA más frecuentes consistieron en erupción cutánea y diarrea (de cualquier grado, 75% y 54%, respectivamente); la mayoría fueron de grados 1 o 2 y controlables sin intervención. Erupción y diarrea de grados 3 o 4 se presentaron en el 9% y el 6%, respectivamente, de los pacientes tratados con Tarceva y condujeron al abandono del tratamiento en el 1% de ellos en cada uno de estos efectos secundarios. La reducción posológica fue necesaria en el 6% y el 1% de los pacientes con erupción y diarrea, respectivamente. En el estudio BR.21, la mediana de tiempo hasta la aparición de erupción fue de 8 días, y de diarrea, de 12 días.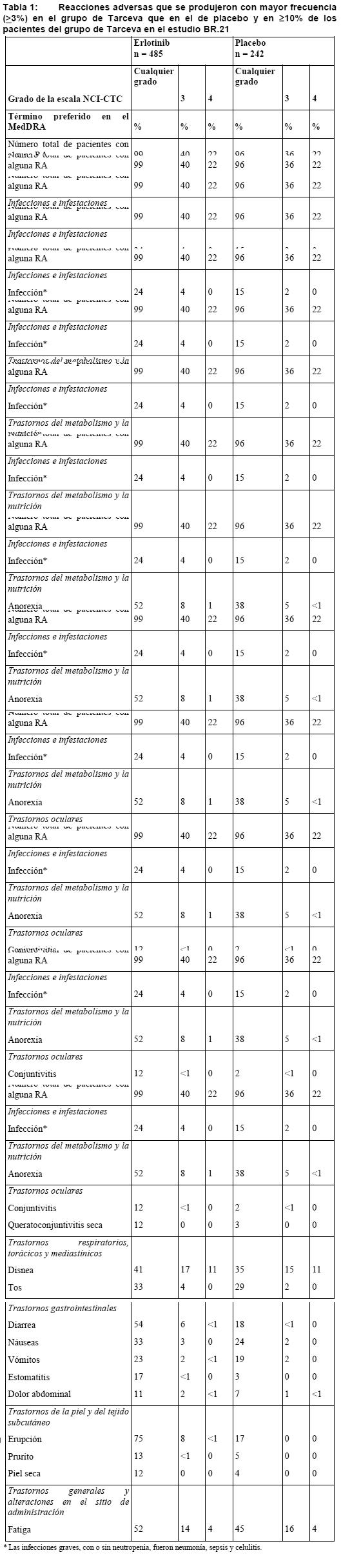
En otro estudio de fase III de doble-ciego, aleatorizado y controlado con placebo (BO18192) en 889 pacientes con CPNM avanzado, recidivante o metastásico tras quimioterapia estándar de primera línea con derivados del platino, no se detectó ningún nuevo signo de toxicidad. Las RA más frecuentes observadas con Tarceva en el estudio BO18192 consistieron en erupción y diarrea (de cualquier grado, 49% y 20%, respectivamente); la mayoría fueron de grados 1 o 2 y controlables sin intervención. Erupción y diarrea de grado 3 se produjeron en el 6% y el 2% de los pacientes, respectivamente. No hubo ningún caso de erupción o diarrea de grado 4. La erupción y la diarrea dieron lugar a la retirada de Tarceva en el 1% y < 1% de los pacientes, respectivamente. La modificación de la dosis (interrupción o reducción) fue necesaria en el 8,3% y el 3% de los pacientes con erupción y diarrea, respectivamente. En un estudio de fase III (ML 20650) abierto y aleatorizado en 154 pacientes se evaluó la seguridad de Tarceva como tratamiento de primera línea en 75 pacientes que presentaban CPNM con mutaciones activadoras de EGFR, sin que se observaran nuevos signos de toxicidad en estos pacientes. Las RA más frecuentes observadas con Tarceva en el estudio ML 20650 consistieron en erupción y diarrea (de cualquier grado, 80% y 57%, respectivamente); la mayoría fueron de grados 1 o 2 y controlables sin intervención. Erupción y diarrea de grado 3 se produjeron en el 9% y el 4% de los pacientes, respectivamente. No hubo ningún caso de erupción o diarrea de grado 4. La erupción y la diarrea dieron lugar a la retirada de Tarceva en el 1% de los pacientes. La modificación de la dosis (interrupción o reducción) fue necesaria en el 11% y el 7% de los pacientes con erupción y diarrea, respectivamente. Tarceva en combinación con quimioterapia: Las RA enumeradas en la tabla siguiente se basan en los datos del grupo de erlotinib de un estudio clínico controlado (PA.3) en el que 259 pacientes con carcinoma pancreático recibieron Tarceva (100 mg) + gemcitabina, y 256, placebo + gemcitabina. Las RA más frecuentes en el estudio PA.3 en el grupo de Tarceva (100 mg) + gemcitabina fueron fatiga, erupción y diarrea. En el grupo de Tarceva + gemcitabina se notificaron erupción y diarrea de grados 3 o 4 en el 5% de los pacientes. La mediana de duración hasta el comienzo de la erupción y la diarrea fue de 10 y 15 días, respectivamente. A causa de la erupción y la diarrea se redujo la dosis en el 2% de los pacientes y se suspendió la administración en hasta el 1% de los pacientes tratados con Tarceva + gemcitabina. En la cohorte (23 pacientes) del grupo de Tarceva (150 mg) + gemcitabina hubo una tasa mayor de ciertas reacciones adversas específicas de clase, incluida la erupción, que requirieron con mayor frecuencia una reducción de la dosis o la retirada de la medicación.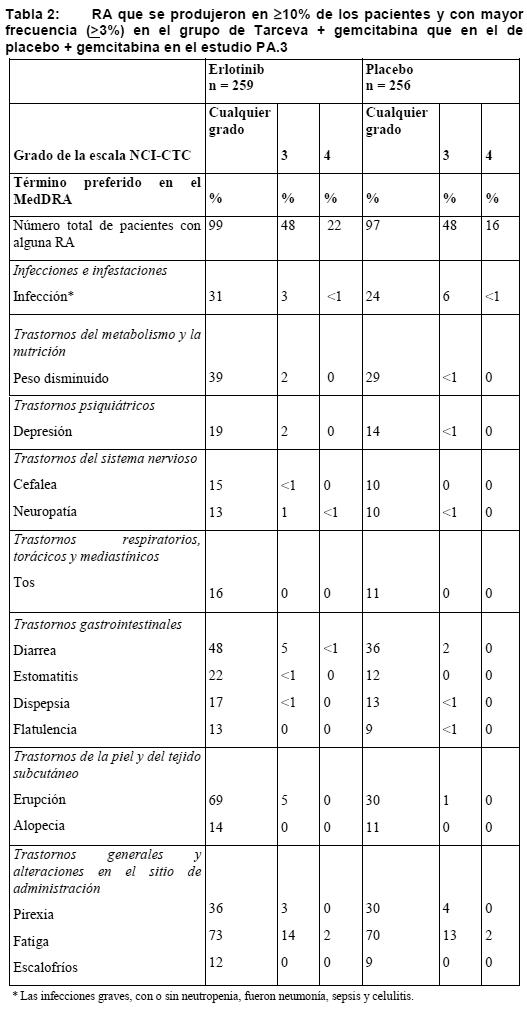
Más información sobre las RA de especial interés: Las RA señaladas a continuación se observaron en pacientes tratados con Tarceva (150 mg) en monoterapia o Tarceva (100 mg o 150 mg) en combinación con gemcitabina. Términos utilizados para clasificar las reacciones adversas por su frecuencia: muy frecuente (≥1/10), frecuente (≥1/100, < 1/10), poco frecuente (≥1/1.000, < 1/100), rara (≥1/10.000, < 1/1000) y muy rara ( < 1/10.000), incluidas notificaciones aisladas. Las Tablas 1 y 2 recogen las RA muy frecuentes; las correspondientes a otras categorías de frecuencia se resumen a continuación. Trastornos gastrointestinales: La perforación gastrointestinal ha sido poco frecuente ( < 1% de los pacientes) en el tratamiento con Tarceva, pero mortal en algunos casos (ver Advertencias). Casos de hemorragia gastrointestinal (algunos mortales) se han notificado con frecuencia, a veces asociados con la administración concomitante de warfarina (ver Interacciones) y/o de AINE. Trastornos hepatobiliares: Valores anómalos en las pruebas funcionales hepáticas (elevación de ALAT, ASAT y bilirrubina) se han descrito con frecuencia en los estudios clínicos de Tarceva. En el estudio PA.3 fueron muy frecuentes. En la mayoría de los casos fueron leves o moderados, transitorios o estaban asociados con metástasis hepáticas. En raras ocasiones se ha notificado insuficiencia hepática (mortal en algún caso) durante el tratamiento con Tarceva. Factores confusores han sido una hepatopatía preexistente o el uso concomitante de medicamentos hepatotóxicos (ver Advertencias). Trastornos oculares: En muy raras ocasiones se ha descrito perforación o ulceración de la córnea en pacientes tratados con Tarceva (ver Advertencias). Queratitis y conjuntivitis se han descrito con frecuencia durante el tratamiento con Tarceva. También se ha descrito crecimiento anómalo de las pestañas (pestañas incarnadas, hipercrecimiento y espesamiento de las pestañas) (ver Advertencias). Trastornos respiratorios, torácicos y mediastínicos: Las notificaciones de episodios similares a la EPI graves (incluido el fallecimiento) en los pacientes tratados con Tarceva contra el CPNM u otros tumores sólidos avanzados han sido poco frecuentes (ver Advertencias). Casos de epistaxis se han descrito frecuentemente en los estudios tanto de CPNM como de carcinoma pancreático. Trastornos de la piel y del tejido subcutáneo: Erupción cutánea se ha notificado con mucha frecuencia en los pacientes tratados con Tarceva. En general, se manifiesta en forma de erupción eritematosa o papulopustulosa leve o moderada, que puede producirse o empeorar en las zonas del cuerpo expuestas al sol. Para los pacientes expuestos al sol puede ser recomendable utilizar vestidos protectores y/o un filtro solar (por ejemplo, uno con minerales). Se han observado casos frecuentes de acné, dermatitis acneiforme y foliculitis, en su mayoría de intensidad leve o moderada y no graves. Fisuras de la piel, la mayor parte de las veces no graves, se han descrito con frecuencia; en la mayoría de los casos estaban asociadas con erupción y piel seca. Otras reacciones cutáneas leves como hiperpigmentación han sido poco frecuentes ( < 1% de los pacientes). Se han descrito trastornos cutáneos ampollosos, vesiculosos y exfoliativos, incluidos muy raros casos evocadores del síndrome de Stevens-Johnson/Lyell (necrólisis epidérmica tóxica), algunos de ellos mortales (ver Advertencias). En los estudios clínicos se han descrito alteraciones del pelo y las uñas, en su mayoría no graves; por ejemplo, paroniquia se ha notificado con frecuencia, mientras que han sido poco frecuentes las descripciones de cambios en las cejas o las pestañas y de uñas quebradizas u onicólisis. Experiencia tras la comercialización: Trastornos de la piel y del tejido subcutáneo: En la farmacovigilancia tras la comercialización han sido poco frecuentes las notificaciones de alteraciones del pelo o las uñas y en su mayoría no graves; por ejemplo, hirsutismo, cambios en las cejas o las pestañas, paroniquia y uñas quebradizas u onicólisis.
Advertencias.
Enfermedad pulmonar intersticial: Casos similares a la enfermedad pulmonar intersticial (EPI) se han descrito con poca frecuencia en pacientes tratados con Tarceva contra CPNM, carcinoma pancreático u otros tumores sólidos avanzados. En el estudio fundamental BR.21 en pacientes con CPNM, la incidencia de episodios similares a la EPI graves fue del 0,8% tanto en el grupo de placebo como en el de Tarceva. En el estudio sobre el carcinoma pancreático en combinación con gemcitabina, la incidencia de episodios similares a la EPI fue del 2,5% en el grupo de Tarceva + gemcitabina frente al 0,4% en el grupo de placebo + gemcitabina. La incidencia global entre los pacientes tratados con Tarceva en todos los estudios (incluidos los realizados sin grupo de control y los estudios con quimioterapia concomitante) ha sido de aproximadamente el 0,6%. Algunos ejemplos de diagnósticos notificados en pacientes con sospecha de episodios similares a la EPI son: neumonitis, neumonitis por irradiación, neumonitis por hipersensibilidad, neumonía intersticial, enfermedad pulmonar intersticial, bronquiolitis obliterante, fibrosis pulmonar, síndrome de dificultad respiratoria aguda, infiltración pulmonar y alveolitis. El comienzo de estos episodios similares a la EPI osciló entre algunos días y varios meses desde el inicio del tratamiento con Tarceva. En la mayoría de los casos se dieron factores que pudieron contribuir o confundir el diagnóstico, como quimioterapia concomitante o previa, radioterapia previa, enfermedad parenquimatosa pulmonar preexistente, enfermedad pulmonar metastásica o infecciones pulmonares. En pacientes con un comienzo agudo de síntomas pulmonares inexplicables, nuevos y/o progresivos, como disnea, tos y fiebre, se debe interrumpir la administración de Tarceva hasta que se realice una evaluación diagnóstica. Si se diagnostica EPI, se debe suspender el tratamiento con Tarceva e iniciar el tratamiento apropiado necesari