TRACLEER COMPRIMIDOS RECUBIERTOS
BIOTOSCANA
Antihipertensivo pulmonar.
Composición.
Cada comprimido recubierto contiene 62,5 o 125 mg de bosentán (monohidrato). Núcleo del comprimido: Almidón de maíz, Almidón pregelatinizado, Glicolato sódico de almidón, Povidona, Dibehenato de glicerol, Estearato de magnesio. Recubrimiento: Hipromelosa Triacetato de glicerol Talco Dióxido de titanio (E171), Oxido de hierro amarillo (E172), Oxido de hierro rojo (E172), Etilcelulosa. Forma farmacéutica: Comprimido recubierto: De forma ovalada, biconvexo, de color naranja-blanco recubierto con película y con 62,5 o 125 grabado en una cara.
Farmacología.
Propiedades farmacodinámicas: Grupo farmacoterapéutico: otros agentes para la hipertensión, código ATC: C02KX01. Mecanismo de acción: Bosentán es un antagonista dual de los receptores de la endotelina (ERA) con afinidad por los receptores de endotelina A y B (ETA y ETB). Bosentán disminuye la resistencia vascular tanto pulmonar como sistémica, dando lugar a un aumento del gasto cardíaco sin aumento de la frecuencia cardiaca. La endotelina-1 (ET-1), una neurohormona, es uno de los vasoconstrictores más potentes conocidos, y también induce fibrosis, proliferación celular, hipertrofia cardiaca, y remodelación, siendo además proinflamatoria. Estos efectos están mediados por la unión de la endotelina a los receptores ETA y ETB situados en las células del músculo liso vascular y el endotelio. Las concentraciones de ET-1 en tejidos y plasma aumentan en distintos trastornos cardiovasculares y enfermedades del tejido conectivo, incluidas la hipertensión arterial pulmonar, esclerodermia, insuficiencia cardiaca aguda y crónica, isquemia miocárdica, hipertensión sistémica y ateroesclerosis, lo cual sugiere un papel patogénico de la ET-1 en estas enfermedades. En la hipertensión arterial pulmonar e insuficiencia cardiaca, en ausencia de un antagonista de los receptores de endotelina, las concentraciones de elevadas de ET-1 están en estrecha correlación con la gravedad y el pronóstico de estas enfermedades. Bosentán compite con la unión de la ET-1 y otros péptidos ET, a ambos receptores ETA y ETB, con una afinidad ligeramente superior por los receptores ETA (Ki = 4,1-43 nM) que por los receptores ETB (KI = 38-730 nM). Bosentán es un antagonista específico de los receptores ET y no se une a otros receptores. Eficacia Modelos animales: En modelos animales de hipertensión pulmonar, la administración oral crónica de bosentán prolongó la supervivencia, redujo la resistencia vascular pulmonar e invirtió la hipertrofia vascular pulmonar y la hipertrofia ventricular derecha. En los modelos animales de fibrosis pulmonar, bosentán redujo el depósito de colágeno en los pulmones. Eficacia en pacientes adultos con hipertensión arterial pulmonar: Se han realizado dos ensayos multicéntricos, aleatorizados, doble ciego, controlados con placebo en 32 (estudio AC-052-351) y 213 (estudio AC-052-352, BREATHE-1) pacientes adultos en clase funcional III-IV de la OMS para la hipertensión arterial pulmonar (hipertensión pulmonar primaria o hipertensión pulmonar secundaria principalmente a esclerodermia). Después de 4 semanas de tratamiento con 62,5 mg de Tracleer dos veces al día, las dosis de mantenimiento estudiadas en estos ensayos fueron de 125 mg dos veces al día en AC-052-351 y 125 mg dos veces al día o 250 mg dos veces al día en AC-052-352. Tracleer fue añadido al tratamiento que en ese momento estuviese recibiendo el paciente, y que incluir una combinación de anticoagulantes, vasodilatadores (p. ej., bloqueantes de los canales del calcio), diuréticos, oxígeno y digoxina, pero no epoprostenol. Como control se empleó placebo además del tratamiento del paciente en ese momento. La variable primaria de eficacia en cada ensayo fue el cambio en la distancia recorrida en la prueba de la marcha de 6 minutos a las 12 semanas para el primer estudio, y a las 16 semanas para el segundo estudio. En ambos ensayos, el tratamiento con Tracleer se tradujo en aumentos significativos de la capacidad de ejercicio. El aumento, ajustado por placebo, en la distancia recorrida comparada con los valores basales fue de 76 metros (p = 0,02; prueba t) y 44 metros (p = 0,0002; prueba U de Mann-Whitney) como variable principal de eficacia en cada ensayo, respectivamente. Las diferencias entre los grupos a los que se administró 125 mg dos veces al día o 250 mg dos veces al día no fueron estadísticamente significativas, pero se observó una tendencia hacia una mejoría de la capacidad de ejercicio en el grupo tratado con 250 mg dos veces al día. La mejoría en la distancia recorrida se observó a las 4 semanas de tratamiento, fue claramente evidente a las de 8 semanas de tratamiento y se mantuvo durante un total de 28 semanas de tratamiento doble ciego en un subconjunto de pacientes. En un análisis retrospectivo de la respuesta, basado en el cambio en la distancia recorrida, la clase funcional OMS y la disnea de los 95 pacientes aleatorizados a recibir Tracleer 125 mg dos veces al día en los ensayos controlados con placebo, se evidenció que a la semana 8, 66 pacientes habían mejorado, 22 estaban estables y 7 habían empeorado. De los 22 pacientes estables en la semana 8, 6 mejoraron en la semana 12/16 y 4 presentaron un deterioro en comparación con los parámetros basales. De los 7 pacientes que habían empeorado en la semana 8, 3 mejoraron en la semana 12/16 y 4 empeoraron respecto a la situación basal. Sólo se evaluaron parámetros hemodinámicos invasivos en el primer estudio. El tratamiento con Tracleer provocó un aumento significativo del índice cardiaco asociado con una reducción significativa de la presión arterial pulmonar, resistencia vascular pulmonar y presión auricular derecha media. Se observó una reducción de los síntomas de hipertensión arterial pulmonar con el tratamiento con Tracleer. La valoración de la disnea durante la prueba de la marcha mostró una mejoría en los pacientes tratados con Tracleer. En el ensayo AC-052-352, el 92% de los 213 pacientes fueron clasificados en fase basal como clase funcional III de la OMS y el 8% como clase IV. El tratamiento con Tracleer resultó en una mejoría de la clase funcional de la OMS en el 42,4% de los pacientes (30,4% en los tratados con placebo). El cambio global en la clase funcional de la OMS durante ambos ensayos fue significativamente superior entre los pacientes tratados con Tracleer comparado con los pacientes tratados con placebo. El tratamiento con Tracleer se asoció a una reducción significativa en la tasa de deterioro clínico a las 28 semanas, comparado con los tratados con placebo (10,7% comparado con 37,1%, respectivamente: p = 0,0015). En un ensayo prospectivo, multicéntrico, aleatorizado, doble ciego, controlado con placebo (BREATHE-5), los pacientes con HAP de clase funcional III de la OMS y fisiología de Eisenmenger asociada a cardiopatía congénita recibieron Tracleer, 62,5 mg dos veces al día durante 4 semanas seguido de 125 mg dos veces al día durante 12 semanas (n = 37) o placebo (n = 17). El objetivo principal fue demostrar que Tracleer no empeora la hipoxemia. Tras 16 semanas, Tracleer aumentó la media de saturación de oxígeno en un 1,0% (IC 95% -0,7; 2,8%) comparado con placebo, demostrando que bosentán no empeora la hipoxemia. El tratamiento con Tracleer produjo una reducción significativa de El valor medio de resistencia vascular pulmonar se redujo significativamente en el grupo tratado con bosentán (el mayor efecto se observó en el subgrupo de pacientes que presentaban cortocircuito intracardiaco bidireccional). Tras 16 semanas, el incremento medio corregido por placebo en la distancia recorrida en la prueba de la marcha de 6 minutos fue de 53 metros (p = 0,0079), reflejando una mejoría de la capacidad de ejercicio. Se realizó un ensayo abierto, no controlado (AC-052-362; BREATHE-4), en 16 pacientes con HAP asociada a infección por VIH en clase funcional III. Los pacientes fueron tratados con Tracleer 62,5 mg dos veces al día durante 4 semanas, seguido de 125 mg dos veces al día durante las 12 semanas siguientes. Después de 16 semanas de tratamiento se observaron mejorías significativas respecto a la situación basal en la capacidad de ejercicio: el aumento medio en la prueba de la marcha de 6 minutos fue de + 91.4 metros con respecto a los 332,6 metros basales de promedio (p < 0,001). Como parte de la evaluación de la seguridad, se valoró el efecto de Bosentán sobre la terapia antirretroviral (es decir, la reducción de las concentraciones sistémicas de los fármacos antirretrovirales coadministrados que tuvieran un impacto negativo en la eficacia del tratamiento antirretroviral) a través del recuento de células CD4 y la titulación del RNA HIV-1. Dadas las limitaciones del estudio (número reducido de pacientes, investigación farmacocinética de fármacos antirretrovirales no estandarizada, heterogeneidad de los regímenes farmacológicos), no se pudieron extraer conclusiones definitivas en relación a los efectos de bosentán sobre la eficacia de los fármacos antirretrovirales. Se observó un aumento de la carga viral en 5 pacientes. No se dispone de datos posteriores a las 16 semanas de este ensayo. No se han realizado ensayos para demostrar los efectos favorables del tratamiento con Tracleer sobre la supervivencia. No obstante, se ha hecho un seguimiento del estado de la totalidad de los 235 pacientes que fueron tratados con bosentán en los dos estudios pivotales controlados con placebo (AC052-351 y AC-052-352) y/o sus dos extensiones, sin tratamiento control, en fase abierta. La duración media de la exposición a bosentán fue de 1,9 ± 0,7 año [mín: 0,1; máx: 3,3 años] y la media de seguimiento fue de 2,0 ± 0,6 años. La mayoría de los pacientes estaban diagnosticados de hipertensión pulmonar primaria (PPH) (72%) y se incluían en la clase funcional III de la OMS (84%). En el conjunto global de esta población, la supervivencia estimada según el método de Kaplan-Meier fue del 93% y 84% para el primer y segundo año desde el inicio del tratamiento, respectivamente. Las estimaciones de supervivencia fueron inferiores en el subgrupo de pacientes con HAP secundaria a esclerosis sistémica. El inicio de tratamiento con epoprostenol puede haber influido en las estimaciones de 43/235 pacientes. Ensayo en niños con hipertensión arterial pulmonar: Se ha realizado un ensayo en niños con hipertensión pulmonar. Tracleer ha sido evaluado en un ensayo abierto, no controlado, en 19 pacientes pediátricos con hipertensión arterial pulmonar (AC052-356, BREATHE-3: hipertensión pulmonar primaria en 10 pacientes e hipertensión arterial pulmonar relacionada a cardiopatía congénita en 9). Este estudio se diseñó principalmente como un estudio farmacocinético. Los pacientes fueron divididos en tres grupos de dosis de acuerdo con tres grupos de pesos corporales y recibieron tratamiento durante 12 semanas. La mitad de los pacientes asignados a cada grupo estaba recibiendo ya epoprostenol por vía intravenosa, esta dosis de epopostrenol se mantuvo constante durante todo el ensayo. Las edades estaban comprendidas entre 3 y 15 años. En el periodo basal, los pacientes se hallaban en clase funcional II (n = 15, 79%) o III (n = 4, 21%) de la OMS. Se obtuvieron parámetros hemodinámicos en 17 pacientes. El aumento medio del índice cardíaco fue de 0,5 l/min/m2, el descenso medio en la presión arterial pulmonar media fue de 8 mmHg y el descenso medio en la resistencia vascular pulmonar de 389 dinas·seg·cm -5. Esta mejoría hemodinámica tuvo lugar tanto con el tratamiento simultáneo con epoprostenol como sin él. Los cambios en los parámetros relacionados con la capacidad de ejercicio en la semana 12, en relación con los parámetros basales, fueron muy variables y ninguno fue significativo. Tratamiento combinado con epoprostenol: La combinación de Tracleer y epoprostenol ha sido investigada en dos ensayos: el AC-052-355 (BREATHE-2) y el AC-052-356 (BREATHE-3). El estudio AC-052-355 fue un ensayo aleatorizado, multicéntrico, doble ciego, en grupos paralelos, en el que se comparó Tracleer con placebo en 33 pacientes con hipertensión arterial pulmonar grave que estaban recibiendo tratamiento con epoprostenol. El estudio AC-052-356, fue un ensayo abierto, no controlado en el que 10 de los 19 pacientes pediátricos recibieron tratamiento concomitante con Tracleer y epoprostenol durante las 12 semanas del ensayo. El perfil de seguridad de la combinación no difirió del esperado para cada uno de los fármacos por separado, siendo la combinación bien tolerada tanto en niños como en adultos. No se ha demostrado el beneficio clínico de la combinación. Esclerosis sistémica con afectación digital ulcerosa activa: Se han realizado dos ensayos clínicos multicéntricos, aleatorizados, doble ciego y controlados con placebo en 122 (Estudio AC-052-401, RAPIDS-1) y 190 (Estudio AC-052-331, RAPIDS-2) en pacientes adultos con esclerosis sistémica con afectación digital ulcerosa activa (tanto úlceras digitales activas como antecedentes de úlceras digitales en el último año). En el estudio AC-052-331, los pacientes debían tener al menos una úlcera digital de reciente aparición, y entre los dos ensayos un 85% de los pacientes tenían afectación digital ulcerosa activa en condiciones basales. Después de 4 semanas de tratamiento con Tracleer 62,5 mg dos veces al día, la dosis de mantenimiento en ambos ensayos clínicos fue de 125 mg dos veces al día. La duración del tratamiento doble ciego fue de 16 semanas en el estudio AC-052-401, y de 24 semanas en el estudio AC-052-331. Se permitió la administración de tratamientos para la esclerosis sistémica y las úlceras digitales si éstos permanecían constantes durante al menos 1 mes antes del inicio del tratamiento y durante el periodo en que en el ensayo era doble ciego. La variable principal en ambos estudios fue el número de nuevas úlceras digitales en comparación con la situación basal. En el grupo tratado con Tracleer aparecieron menos úlceras digitales nuevas durante la duración del tratamiento en comparación con placebo. Durante las 16 semanas de tratamiento dobleciego en el ensayo AC-052-401, los pacientes desarrollaron una media de 1,4 nueva úlcera digital en el grupo de bosentán vs. 2,7 nuevas úlceras digitales en el grupo placebo (p = 0,0042). Durante las 24 semanas de tratamiento doble ciego en el estudio AC-052-331, las cifras correspondientes fueron de 1,9 vs. 2,7 nuevas úlceras digitales, respectivamente (p = 0,0351). En ambos estudios los pacientes que recibían bosentán tuvieron menor probabilidad de desarrollar múltiples nuevas úlceras digitales durante el ensayo y tardaron más tiempo en desarrollar nuevas úlceras digitales, en comparación con los pacientes que recibían placebo. El efecto de bosentán en la reducción del número de nuevas úlceras digitales fue más pronunciado en los pacientes con úlceras digitales múltiples. No se observó ningún efecto de bosentán en el tiempo hasta la curación de las úlceras digitales en ninguno de los dos ensayos. Propiedades farmacocinéticas: La farmacocinética de bosentán ha sido estudiada principalmente en sujetos sanos. Los datos, de carácter limitado, obtenidos en pacientes, demuestran que la exposición a bosentán en pacientes adultos con hipertensión arterial pulmonar es unas 2 veces superior a la observada en sujetos adultos sanos. En adultos sanos, bosentán muestra una farmacocinética dosis- y tiempo-dependiente. El aclaramiento y el volumen de distribución disminuyen con dosis intravenosas crecientes y aumentan con el tiempo. Después de la administración oral, la exposición sistémica es proporcional a la dosis hasta los 500 mg. A dosis orales más elevadas, Cmáx y AUC aumentan menos que en proporción a la dosis. Absorción: En voluntarios sanos, la biodisponibilidad absoluta de bosentán es aproximadamente del 50%, y no se ve afectada por los alimentos. Las concentraciones plasmáticas máximas se alcanzan en un período de 3 a 5 horas. Distribución: Bosentán se fija en gran medida a las proteínas plasmáticas ( > 98%), principalmente a la albúmina. Bosentán no penetra en los eritrocitos. Tras una dosis intravenosa de 250 mg se determinó un volumen de distribución (Vss) de unos 18 litros. Biotransformación y eliminación: Después de una dosis intravenosa única de 250 mg, el aclaramiento fue de 8,2 l/h. La vida media de eliminación terminal (t1/2) es de 5,4 horas. Después de la administración de dosis múltiples, las concentraciones plasmáticas de bosentán disminuyen gradualmente al 50%-65% de las observadas después de la administración de dosis únicas. Esta disminución probablemente se debe a la autoinducción de las enzimas hepáticas involucradas en su metabolismo. El equilibrio estacionario se alcanza en un plazo de 3 a 5 días. Bosentán es eliminado por excreción biliar después de su metabolismo hepático por las isoenzimas CYP3A4 y CYP2C9 del citocromo P450. Menos del 3% de la dosis oral administrada se recupera en la orina. Bosentán forma tres metabolitos, de los cuales sólo uno es farmacológicamente activo. Este metabolito se excreta principalmente inalterado por vía biliar. En pacientes adultos, la exposición al metabolito activo es mayor que en sujetos sanos. La exposición al metabolito activo puede estar incrementada en pacientes con evidencia de colestasis. Bosentán es un inductor del CYP3A4 y CYP2C9 y posiblemente también del CYP2C19 y la P-glucoproteína. In vitro, bosentán inhibe la bomba exportadora de sales biliares en cultivos de hepatocitos. Datos in vitro han demostrado que bosentán no ejerce ningún efecto inhibidor significativo sobre las isoenzimas del CYP ensayadas (CYP1A2, 2A6, 2B6, 2C8, 2C9, 2D6, 2E1, 3A4). Por consiguiente, no se espera que el bosentán aumente las concentraciones plasmáticas de fármacos metabolizados por estas isoenzimas. Farmacocinética en poblaciones especiales: En relación con el intervalo estudiado para cada variable, no se espera que la farmacocinética de bosentán en adultos se vea influida de forma relevante por el sexo, peso corporal, raza o edad. No se dispone de datos farmacocinéticos en niños menores de 3 años. Niños: La farmacocinética en dosis orales únicas y múltiples ha sido estudiada en pacientes pediátricos con hipertensión arterial pulmonar cuya dosis se había ajustado de acuerdo con el peso corporal. La exposición a bosentán disminuyó con el tiempo en concordancia con las conocidas propiedades autoinductoras enzimáticas de bosentán. Los valores medios de AUC (CV%) de bosentán en pacientes pediátricos tratados con dosis de 31,25; 62,5 ó 125 mg b.i.d. fueron de 3496 (49), 5428 (79) y 6124 (27) ng·h/ml, respectivamente, y fueron menores que el valor de 8149 (47) ng·h/ml observado en pacientes adultos con hipertensión arterial pulmonar que recibieron dosis de 125 mg b.i.d. En el estado estacionario, la exposición sistémica en pacientes pediátricos con pesos entre 10 y 20 kg, 20 y 40 kg y más de 40 kg fue del 43%, 67% y 75% de la observada en pacientes adultos, respectivamente. No está clara la razón de esta diferencia. Probablemente sea debida a un mayor metabolismo hepático así como a una mayor excreción. Se desconocen las consecuencias de estos hallazgos sobre una posible hepatotoxicidad. Ni el sexo, ni el uso concomitante de epoprostenol por vía intravenosa, mostraron un efecto importante sobre la farmacocinética de bosentán. Insuficiencia hepática En pacientes con insuficiencia hepática leve (Child-Pugh clase A) no se observaron cambios relevantes en la farmacocinética. En el estado estacionario el AUC de bosentán y el AUC del metabolito activo, Ro 48-5033, fueron un 9% y un 33% más altos, respectivamente, en pacientes con insuficiencia hepática leve comparados con los valores observados en voluntarios sanos. No se ha estudiado la farmacocinética de bosentán en pacientes con insuficiencia hepática clase B o C de Child-Pugh por lo que Tracleer está contraindicado en esta población. Insuficiencia renal: En pacientes con insuficiencia renal grave (aclaramiento de creatinina de 15-30 ml/min), las concentraciones plasmáticas de bosentán disminuyeron en aproximadamente un 10%. Las concentraciones plasmáticas de los metabolitos de bosentán fueron aproximadamente dos veces superiores en estos pacientes en comparación con los valores en voluntarios con función renal normal. No se requiere ajuste de dosis en pacientes con insuficiencia renal. No se tiene experiencia clínica específica en pacientes sometidos a diálisis. Dadas las propiedades fisicoquímicas y el alto nivel de fijación a proteínas, no se espera que bosentán sea eliminado de la circulación de forma significativa mediante diálisis. Datos preclínicos sobre seguridad: Un estudio de carcinogénesis a 2 años en ratones mostró una mayor incidencia combinada de carcinomas y adenomas hepatocelulares en machos, pero no en hembras, a concentraciones plasmáticas entre 2 a 4 veces las concentraciones plasmáticas alcanzadas a dosis terapéuticas en el ser humano. En ratas, la administración oral de bosentán durante 2 años produjo un pequeño aumento significativo en la incidencia combinada de carcinomas y adenomas de células foliculares de tiroides en machos, pero no en hembras, a concentraciones plasmáticas de entre 9 a 14 veces las concentraciones plasmáticas alcanzadas a dosis terapéuticas en el ser humano. Las pruebas de genotoxicidad con bosentán fueron negativas. En ratas, se observó evidencia de discretas alteraciones hormonales tiroideas inducidas por bosentán. Sin embargo, no hubo evidencia de que bosentán afectara la función tiroidea (tiroxina, TSH) en el ser humano. Se desconoce el efecto de bosentán sobre la función mitocondrial. Bosentán ha demostrado ser teratógeno en ratas a niveles plasmáticos superiores a 1,5 vez las concentraciones plasmáticas alcanzadas a las dosis terapéuticas en humanos. Los efectos teratógenos, entre los que se incluyen malformaciones de cabeza, cara y grandes vasos, fueron dosis-dependientes. La similitud del patrón de malformaciones observadas con otros antagonistas de los receptores de la ET, así como en ratones carentes de ET indica un efecto de clase. Deberán adoptarse las precauciones adecuadas en mujeres en edad fértil. En estudios de fertilidad en ratas macho y hembra a concentraciones plasmáticas entre 21 y 43 veces, respectivamente, las esperadas a dosis terapéuticas en humanos, no se observaron efectos en el recuento, movilidad y viabilidad de los espermatozoides, ni sobre el apareamiento o fertilidad, así como tampoco se observaron efectos adversos en el desarrollo del embrión previo a la implantación o sobre la implantación.
Indicaciones.
Tratamiento de la hipertensión arterial pulmonar (HAP) para mejorar los síntomas y la capacidad de ejercicio en pacientes con grado funcional III. Se ha demostrado eficacia en: HAP primaria (idiopática y familiar), HAP secundaria a la esclerodermia sin enfermedad pulmonar intersticial significativa, HAP asociada a cortocircuitos sistémico-pulmonares congénitos y fisiología de Eisenmenger, Tracleer también está indicado para la reducción del número de nuevas úlceras digitales en pacientes con esclerosis sistémica con alteración digital ulcerosa.
Dosificación.
Hipertensión Arterial Pulmonar: El tratamiento sólo debe ser iniciado y controlado por un médico experimentado en el tratamiento de la hipertensión arterial pulmonar. El tratamiento con Tracleer se iniciará a dosis de 62,5 mg dos veces al día durante 4 semanas, aumentando entonces la dosis a 125 mg, dos veces al día (dosis de mantenimiento). Los comprimidos se administrarán por vía oral por la mañana y por la noche, con o sin alimentos. En el caso de deterioro clínico (p. ej., reducción de al menos un 10 % en la distancia recorrida en la prueba de la marcha de 6 minutos en comparación con la determinación previa al tratamiento) pese al tratamiento con Tracleer durante al menos 8 semanas (por lo menos cuatro semanas con dosis de mantenimiento), debe considerarse el empleo de tratamientos alternativos. No obstante, algunos pacientes que no respondan al tratamiento con Tracleer después de 8 semanas, pueden responder de manera favorable después de 4 a 8 semanas adicionales de tratamiento. Si se decide retirar el tratamiento con Tracleer, deberá hacerse de manera paulatina mientras se introduce un tratamiento alternativo. En el caso de empeoramiento clínico tardío pese al tratamiento con Tracleer (es decir, después de varios meses de tratamiento), este tratamiento deberá ser evaluado de nuevo. Ciertos pacientes que no responden bien a 125 mg de Tracleer dos veces al día pueden mejorar ligeramente su capacidad de ejercicio cuando la dosis se aumenta a 250 mg dos veces al día. Deberá realizarse una cuidadosa evaluación del balance riesgo/beneficio, teniendo en cuenta que la toxicidad hepática es dosisdependiente. Interrupción del tratamiento: La experiencia en relación con la interrupción brusca de Tracleer es limitada. No se ha observado evidencia de efecto rebote. Sin embargo, para evitar la aparición de un posible deterioro clínico contraproducente debido a un potencial efecto rebote, debe considerarse la reducción paulatina de la dosis (reduciendo esta a la mitad durante 3 a 7 días). Se recomienda intensificar la vigilancia durante el periodo de interrupción. Esclerosis Sistémica con afectación digital ulcerosa activa: El tratamiento sólo debe ser iniciado y controlado por un médico experimentado en el tratamiento de la esclerosis sistémica. El tratamiento con Tracleer se iniciará a dosis de 62,5 mg dos veces al día durante 4 semanas, aumentando entonces la dosis a 125 mg, dos veces al día. Los comprimidos se administrarán por vía oral por la mañana y por la noche, con o sin alimentos. La experiencia en ensayos clínicos controlados para esta indicación se limita a 6 meses. La respuesta al tratamiento y la necesidad de terapia continuada deberá ser reevaluada regularmente. Deberá realizarse una adecuada evaluación de la relación beneficio/riesgo, teniendo en cuenta la toxicidad hepática de Bosentan. Poblaciones especiales: Pauta de dosificación en insuficiencia hepática: No es necesario ajustar la dosis en pacientes con insuficiencia hepática leve (es decir, Child-Pugh clase A). Tracleer está contraindicado en pacientes con insuficiencia hepática de moderada a severa. Pauta de dosificación en insuficiencia renal: No es necesario ajustar la dosis en pacientes con insuficiencia renal. No se requiere ajustar la dosis en pacientes sometidos a diálisis. Pauta de dosificación en ancianos: No es necesario ajustar la dosis en pacientes mayores de 65 años. Niños -Hipertensión Arterial Pulmonar: La seguridad y eficacia en pacientes menores de 12 años no está plenamente documentada. La siguiente pauta de tratamiento se empleó en el estudio AC-052-356 (BREATHE-3):
Este ensayo fue diseñado principalmente para estudiar la farmacocinética en niños. El número de pacientes estudiado en cada grupo de dosis fue insuficiente para establecer la pauta de tratamiento óptima en pacientes menores de 12 años. Los datos farmacocinéticos obtenidos mostraron que la exposición sistémica era menor que la observada en pacientes adultos con hipertensión pulmonar por lo que el efecto sobre el árbol vascular pulmonar puede ser insuficiente. Sin embargo, no se ha establecido la seguridad de dosis más elevadas en niños. No hay experiencia en niños menores de 3 años. Esclerosis sistémica con afectación digital ulcerosa activa: No se disponen de datos de seguridad y eficacia en pacientes menores de 18 años. Pacientes con bajo peso corporal: La experiencia en pacientes con peso corporal inferior a 40 kg es limitada.
Contraindicaciones.
Hipersensibilidad al principio activo o a alguno de los excipientes Child-Pugh Clase B o C, es decir, insuficiencia hepática de moderada a grave. Valores basales de aminotransferasas hepáticas, es decir, aspartato aminotransferasa (AST) y/o alanina aminotransferasa (ALT), superiores a 3 veces el límite superior de la normalidad. Empleo concomitante de ciclosporina A. Embarazo Mujeres en edad fértil que no utilicen un método anticonceptivo fiable.
Reacciones adversas.
Hallazgos procedentes de ensayos controlados con placebo: En ocho ensayos controlados con placebo, seis de los cuales fueron en indicaciones no relacionadas con hipertensión arterial pulmonar, un total de 677 pacientes recibieron tratamiento con bosentán a una dosis diaria que osciló entre los 100 mg y los 2000 mg, y 288 pacientes fueron tratados con placebo. La duración del tratamiento osciló entre 2 semanas y 6 meses. Las reacciones adversas que aparecieron con mayor frecuencia en los pacientes a los que se administró bosentán (≥ 3% en los pacientes tratados con bosentán, con una diferencia de ≥ 2%) que en los que recibieron placebo fueron cefalea(15,8% vs. 12,8%), rubor facial (6,6% vs. 1,7%), trastornos de la función hepática (5,9% vs. 2,1%), edema de extremidades inferiores (4,7% vs. 1,4%) y anemia (3,4% vs. 1,0%), y todas ellas fueron dosis-dependientes. Ensayos controlados con placebo en HAP primaria (idiopática/familiar) y en HAP asociada a enfermedades del tejido conjuntivo En la tabla siguiente se resumen las reacciones adversas ocurridas en ≥ 3% de los pacientes tratados con Tracleer (125 y 250 mg dos veces al día) en ensayos controlados con placebo en hipertensión arterial pulmonar, y que fueron más frecuentes en estos pacientes: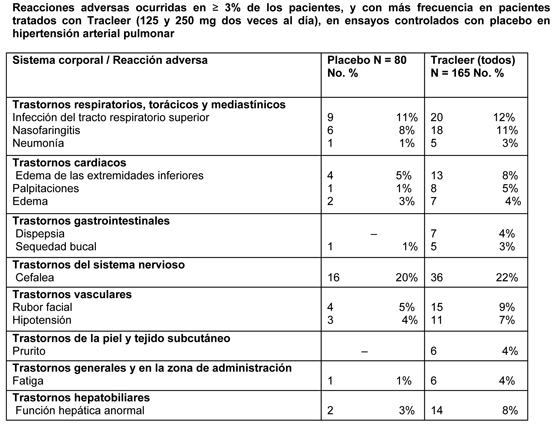
Nota: solamente se incluyen las reacciones adversas que aparecieron desde el comienzo del tratamiento hasta 1 día de calendario después de finalizar el tratamiento. Un paciente puede presentar más de una reacción adversa. A la dosis de mantenimiento recomendada, o doble de la misma (es decir, 125 ó 250 mg dos veces al día), las reacciones adversas que se produjeron con más frecuencia con Tracleer que con placebo (≥ 3% de los pacientes tratados con Tracleer, con una diferencia ≥ 2%) fueron nasofaringitis, rubor facial, función hepática anormal, edema de extremidades inferiores, hipotensión, palpitaciones, dispepsia, fatiga y prurito. Las reacciones adversas que se produjeron en ≥ 1% y < 3% de estos pacientes, y con más frecuencia con Tracleer que con placebo (diferencia de ≥ 2%) fueron anemia, reflujo gastroesofágico y hemorragia rectal, todos en un 2,4% con Tracleer comparado con un 0% con placebo. Las interrupciones del tratamiento debidas a reacciones adversas, durante los ensayos clínicos en pacientes con hipertensión arterial pulmonar, a dosis de 125 y 250 mg dos veces al día, fueron menos frecuentes en los pacientes tratados con Tracleer que en los tratados con placebo (5,5% vs. 10%, respectivamente). Ensayo clínico controlado con placebo en HAP asociada a cardiopatías congénitas (BREATHE-5): El perfil de seguridad de Tracleer en esta población fue similar al observado en los ensayos pivotes en pacientes con HAP. Las reacciones adversas que tuvieron lugar en mayor proporción en pacientes tratados con Tracleer,62,5 mg dos veces al día durante cuatro semanas, seguido por 125 mg dos veces al día (n = 37), que en aquellos que recibieron placebo (n = 17) fueron edema periférico (18,9% vs. 5,9%), cefalea (13,5% vs. 11,8%), palpitaciones (10,8% vs. 0%), mareos (8,1% vs. 5,9%) y dolor torácico (8,1% vs. 0%). Cuatro pacientes interrumpieron el tratamiento debido a reacciones adversas, dos (5,4%) en el grupo bosentán y dos (11,8%) en el grupo placebo. Estudio no controlado en pacientes con HAP asociada a infección por el VIH (BREATHE-4): El perfil de seguridad en esta población (n = 16) cuando fue tratada con Tracleer 62,5 mg dos veces al día durante cuatro semanas, seguido por 125 mg dos veces al día, fue similar al observado en los ensayos clínicos pivotes en pacientes con HAP. Las reacciones adversas más frecuentes fueron el edema periférico (31%), cefalea (19%), alteración de la función hepática (13%), calambres musculares (13%), retención de líquidos (13%) y vómitos (13%). En algunos pacientes se observaron alteraciones hematológicas (anemia y disminución del número de neutrófilos). Ensayos controlados con placebo en úlceras digitales: En la tabla siguiente se resumen las reacciones adversas ocurridas en ≥ 3% de los pacientes tratados con Tracleer (125 mg dos veces al día) en los dos ensayos fundamentales controlados con placebo en úlceras digitales, y que fueron más frecuentes en los pacientes tratados con Tracleer: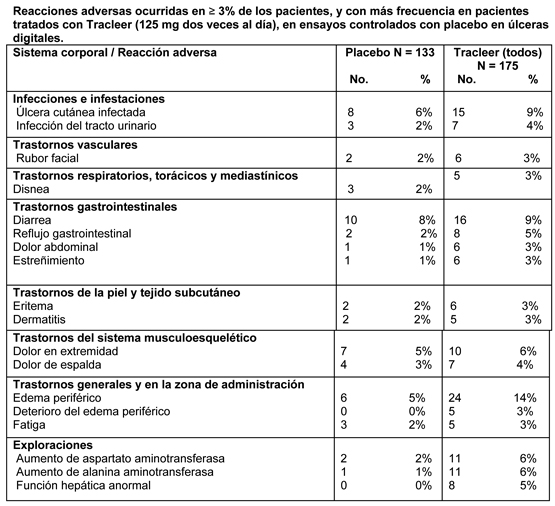
Nota: solamente se incluyen reacciones adversas que aparecieron desde el comienzo del tratamiento hasta 1 día de calendario después de finalizar el tratamiento. Un paciente puede presentar más de una reacción adversa. Alteraciones analíticas: Alteraciones analíticas hepáticas: El empleo de bosentán se ha asociado con una elevación dosis-dependiente de las aminotransferasas hepáticas, es decir, aspartato aminotransferasa y alanina aminotransferasa. Durante el desarrollo clínico, los cambios en los enzimas hepáticos se produjeron generalmente en las 26 primeras semanas del tratamiento y normalmente se desarrollaron de manera paulatina y fueron en su mayoría asintomáticos. En todos los casos los niveles regresaron a los valores previos al tratamiento, sin secuelas, entre pocos días y 9 semanas, bien de manera espontánea o tras reducir la dosis o interrumpir el tratamiento. En el período postcomercialización, se han notificado casos raros de cirrosis hepática e insuficiencia hepática. No está claro el mecanismo por el que aparece este efecto adverso. Estos aumentos en las aminotransferasas pueden revertir espontáneamente mientras se continúa el tratamiento con la dosis de mantenimiento de Tracleer o después de reducir la dosis, aunque puede ser necesaria la interrupción o suspensión del tratamiento. En los ocho ensayos controlados con placebo, seis de los cuales se realizaron en indicaciones distintas a la hipertensión arterial pulmonar, se observaron elevaciones de los niveles de las aminotransferasas hepáticas en más de 3 veces el límite superior de la normalidad (ULN) en el 11,2% de los pacientes tratados con bosentán, comparado con el 1,8% de los pacientes tratados con placebo. En 2 de los 658 (0,3%) pacientes tratados con bosentán se observaron elevaciones de bilirrubina > 3 × ULN asociadas con elevación de las aminotransferasas ( > 3 × ULN). Nueve de los 74 pacientes tratados con bosentán que mostraban elevaciones de las aminotransferasas hepáticas ( > 3 × ULN), también mostraban síntomas tales como dolor abdominal, náuseas/vómitos y fiebre. En ensayos en pacientes con hipertensión arterial pulmonar, la incidencia de elevaciones de las aminotransferasas hepáticas ( > 3 × ULN) fue del 12,7% en los tratados con bosentán (n = 165), del 11,6% en los tratados con 125 mg dos veces al día y del 14,3% en los tratados con 250 mg dos veces al día. Se observaron elevaciones de ocho veces los límites superiores de la normalidad en el 2,1% de pacientes con hipertensión arterial pulmonar tratados con 125 mg dos veces al día y en el 7,1% de pacientes con hipertensión arterial pulmonar tratados con 250 mg dos veces al día. En los dos estudios en pacientes con úlceras digitales la incidencia de elevaciones de las aminotransferasas hepáticas ( > 3 x LSN) fue de 11.3 % en los pacientes tratados con bosentán (N=168) comparado con el 0.8% en los pacientes tratados con placebo (N=129). Se observaron elevaciones > a 8 x LSN en el 2.4 % de los pacientes con úlceras digitales tratados con bosentán. Hemoglobina: El descenso medio en la concentración de hemoglobina desde el período basal hasta la finalización del ensayo en pacientes tratados con bosentán fue de 0,9 g/dl, siendo de 0,1 g/dl en los tratados con placebo. En ocho ensayos controlados con placebo se observó un descenso clínicamente relevante en la hemoglobina (descenso > 15% con respecto los valores basales y valor resultante < 11 g/dl) en el 5,6% de los pacientes tratados con bosentán, comparado con el 2,6% en los pacientes tratados con placebo. En pacientes con hipertensión arterial pulmonar tratados con dosis de 125 y 250 mg dos veces al día, se observaron descensos clínicamente relevantes de la hemoglobina en el 3,0% y 1,3% de los pacientes tratados con bosentán y placebo, respectivamente. En los dos ensayos en pacientes con úlceras digitales, se observó una disminución de los niveles de hemoglobina clínicamente significativa (disminución desde el valor basal que produjo valores de hemoglobina < 10 g/dl) en 4,2 % de los pacientes tratados con bosentán (N=167) comparado con el 3,1% en los pacientes tratados con placebo (N=129). En el periodo post comercialización, se han notificado casos de anemia que han requerido transfusión de glóbulos rojos. Experiencia postcomercialización: La mayoría de las reacciones adversas notificadas durante el periodo postcomercialización han sido similares a las notificadas durante los ensayos clínicos. Las reacciones adversas se agrupan según su frecuencia, de acuerdo con la siguiente convención: muy frecuentes (≥ 1/10); frecuentes (≥ 1/100, < 1/10); poco frecuentes (≥ 1/1.000, < 1/100); raras (≥ 1/10.000, < ≤ 1/1.000); muy raras ( < 1/10.000). Trastornos gastrointestinales: Frecuentes: náuseas. Poco frecuentes: vómitos, dolor abdominal, diarrea. Trastornos hepatobiliares: Poco frecuentes: elevación de aminotransferasas asociada a hepatitis y/o ictericia. Raras: cirrosis hepática, insuficiencia hepática Trastornos de la piel y del tejido subcutáneo: Poco frecuentes: reacciones dehipersensibilidad, incluidas dermatitis, prurito y erupciones cutáneas. Sistema inmunológico: Raras: anafilaxis y/o angioedema. Trastornos de la sangre y del sistema linfático: Frecuentes: Anemia o disminución de hemoglobina, requiriendo alguna vez de transfusión de células rojas. Poco frecuentes: trombocitopenia. En el período postcomercialización se han notificado casos raros de cirrosis hepática de etiología desconocida después de terapia prolongada con Tracleer, en pacientes con múltiples comorbilidades y tratamientos farmacológicos. También se han notificado casos raros de insuficiencia hepática. Estos casos refuerzan la importancia de realizar un estricto cumplimiento del programa mensual de monitorización de la función hepática durante el tratamiento con Tracleer.
Advertencias.
La eficacia de Tracleer no ha sido establecida en pacientes con hipertensión arterial pulmonar grave. Deberá considerarse el cambio a un tratamiento que esté recomendado en la fase grave de la enfermedad (ej., epoprostenol) si empeora la condición clínica. No se ha establecido el balance riesgo/beneficio de bosentán en pacientes en clase funcional I o II de la OMS para la hipertensión arterial pulmonar. El tratamiento con Tracleer sólo deberá iniciarse si la presión arterial sistólica sistémica es superior a 85 mmHg. No se ha observado que Tracleer posea un efecto beneficioso en la curación de las úlceras digitales existentes. Función hepática La elevación de los valores de aminotransferasas hepáticas, es decir, aspartato aminotransferasa y alanina aminotransferasa (AST y/o ALT), asociada con bosentán, es dosis-dependiente. Los cambios en los enzimas hepáticos se producen generalmente durante las primeras 26 primeras semanas del tratamiento aunque también podrían presentarse más tarde. Dicha elevación puede deberse en parte a la inhibición competitiva de la eliminación de las sales biliares de l
