TRUVADA
GADOR CHILE
Antirretroviral. Tratamiento del VIH-SIDA.
Descripción.
Los comprimidos de TRUVADA son comprimidos combinados de dosis fija que contienen emtricitabina y tenofovir disoproxil fumarato. La emtricitabina es un análogo nucleósido sintético de la citidina. El tenofovir disoproxil fumarato (VIREAD, también denominado tenofovir DF) se convierte in vivo en tenofovir, un análogo nucleósido fosfonato acíclico (nucleótido) de adenosina 5'-monofosfato. Tanto la emtricitabina como el tenofovir exhiben una actividad inhibitoria con respecto a la transcriptasa inversa del VIH-1. Los comprimidos de TRUVADA son de administración oral. Cada comprimido, que posee película de recubrimiento, contiene 200mg de emtricitabina y 300mg de tenofovir disoproxil fumarato (equivalente a 245mg de tenofovir disoproxil) como principios activos.
Propiedades.
Microbiología: mecanismo de acción: emtricitabina: la emtricitabina, un análogo nucleósido sintético de la citidina, es fosforilada con enzimas celulares para formar emtricitabina 5'-trifosfato. Emtricitabina 5'-trifosfato inhibe la actividad de la transcriptasa inversa del VIH-1 compitiendo con el sustrato natural de desoxicitidina 5'-trifosfato y al ser incorporada al ADN viral naciente que resulta en la terminación de la cadena. Emtricitabina 5'-trifosfato es una inhibidor débil de las polimerasas a, b, e del ADN de los mamíferos y de la polimerasa g del ADN mitocondrial. Tenofovir disoproxil fumarato: el tenofovir disoproxil fumarato es un análogo nucleósido fosfonato diéster acíclico del monofosfato de adenosina. El tenofovir disoproxil fumarato requiere una hidrólisis inicial del diéster para convertirse en tenofovir y subsiguientes fosforilaciones mediante enzimas celulares para formar el tenofovir difosfato. El tenofovir difosfato inhibe la actividad de la transcriptasa inversa del VIH-1 al competir con el sustrato natural deoxiadenosina 5'-trifosfato y luego de su incorporación al ADN, mediante la terminación de la cadena de ADN. Tenofovir difosfato es un inhibidor débil de las polimerasas a, b del ADN de los mamíferos y de la polimerasa g del ADN mitocondrial. Actividad antiviral: emtricitabina y tenofovir disoproxil fumarato: en estudios de combinación que evaluaban la actividad antiviral in vitro de la emtricitabina y el tenofovir en conjunto, se observaron efectos antivirales sinergísticos. Emtricitabina: la actividad antiviral in vitro de la emtricitabina con respecto a aislados de laboratorio y aislados clínicos de VIH se evaluó en líneas de células linfoblastoideas, en la línea de células MAGI-CCR5 y en células mononucleares sanguíneas periféricas. Los valores IC50 para la emtricitabina estuvieron dentro del rango de 0,0013-0,64mM (0,0003-0,158mg/ml). En estudios de combinación de drogas sobre la emtricitabina con inhibidores nucleósidos de la transcriptasa inversa (abacavir, lamivudina, estavudina, zalcitabina, zidovudina), inhibidores no nucleósidos de la transcriptasa inversa (delavirdina, efavirenz, nevirapina) e inhibidores de la proteasa (amprenavir, nelfinavir, ritonavir, saquinavir), se observaron aditivos a los efectos sinergísticos. La mayoría de estas combinaciones de drogas no se estudiaron en humanos. Emtricitabina presentó una actividad antiviral in vitro con respecto a los clades A, B, C, D, E, F y G del VIH-1 (los valores IC50 oscilaron entre 0,007-0,075mM) y mostraron una actividad específica de las cepas respecto del VIH-2 (los valores IC50 oscilaron entre 0,007-1,5mM). Tenofovir disoproxil fumarato: la actividad antiviral in vitro del tenofovir respecto de aislados de laboratorio y aislados clínicos de VIH-1 se evaluó en líneas de células linfoblastoideas, en células primarias monocito/macrófago y en linfocitos sanguíneos periféricos. Los valores IC50 (50% de concentración inhibitoria) para el tenofovir estuvieron dentro del rango de 0,04-8,5mM. En estudios de combinación de drogas sobre el tenofovir con inhibidores nucleósidos de la transcriptasa inversa (abacavir, didanosina, lamivudina, estavudina, zalcitabina, zidovudina), inhibidores no nucleósidos de la transcriptasa inversa (delavirdina, efavirenz, nevirapina), e inhibidores de la proteasa (amprenavir, indinavir, nelfinavir, ritonavir, saquinavir), se observaron aditivos a los efectos sinergísticos. Tenofovir presentó una actividad antiviral in vitro respecto de los clades A, B, C, D, E, F, G y O del VIH-1 (los valores IC50 oscilaron entre 0,5-2,2mM). Los valores IC50 de tenofovir respecto del VIH-2 oscilaron entre 1,6mM y 4,9mM. Resistencia: emtricitabina y tenofovir disoproxil fumarato: los aislados de VIH-1 con una susceptibilidad reducida a la combinación de emtricitabina y tenofovir se seleccionaron in vitro. El análisis genotípico de estos aislados identificó las sustituciones de aminoácido M184I/V y/o K65R en la transcriptasa inversa viral. Emtricitabina: los aislados del VIH resistentes a la emtricitabina se seleccionaron in vitro. El análisis genotípico de estos aislados señaló que la susceptibilidad reducida a la emtricitabina estaba asociada a una mutación del gen de la transcriptasa inversa del VIH en el codón 184 que resultó en una sustitución de aminoácido de metionina por valina o isoleucina (M184V/I). Los aislados del VIH resistentes a la emtricitabina fueron extraídos de algunos pacientes tratados con emtricitabina sola o en combinación con otros agentes antirretrovirales. En un estudio clínico, los aislados virales de 6/16 (37,5%) de pacientes que no han recibido tratamiento previo con fracaso virológico mostraron una susceptibilidad a la emtricitabina reducida en > 20 veces. El análisis genotípico de estos aislados indicó que la resistencia se debía a las mutaciones M184V/I en el gen de la transcriptasa inversa del VIH. Tenofovir disoproxil fumarato: los aislados de VIH-1 con una susceptibilidad reducida al tenofovir se seleccionaron in vitro. Estos virus manifestaron una mutación K65R en la transcriptasa inversa y señalaron una reducción de 2 a 4 veces en la susceptibilidad al tenofovir. Los aislados de VIH-1 resistentes al tenofovir también fueron extraídos de algunos pacientes tratados con VIREAD en combinación con ciertos agentes antirretrovirales. En pacientes que no han recibido tratamiento previo, 8/47 (17%) de los aislados de los pacientes con un fracaso de VIREAD + lamivudina + efavirenz en la semana 144 indicaron una susceptibilidad in vitro al tenovofir reducida en > 1,4 vez (mediana 3,7). En pacientes que han recibido tratamiento previo, 14/304 (5%, Estudios 902 y 907) de los aislados de los pacientes con fracaso de VIREAD en la semana 96 señalaron una susceptibilidad in vitro al tenovofir reducida en > 1,4 vez (mediana 2,7). El análisis genotípico de los aislados resistentes indicó una mutación en el gen de la transcriptasa inversa del VIH-1 que resultó en la sustitución de aminoácido K65R. Resistencia cruzada: emtricitabina y tenofovir disoproxil fumarato: se ha reconocido la resistencia cruzada entre ciertos inhibidores nucleósidos de la transcriptasa inversa (NRTIs). Las sustituciones M184V/I y/o K65R seleccionadas in vitro por medio de la combinación de emtricitabina y tenofovir también se observaron en algunos aislados de VIH-1 en pacientes que fracasaron en el tratamiento con tenofovir en combinación con lamivudina o emtricitabina, y abacavir o didanosina. Por lo tanto, la resistencia cruzada entre estas drogas puede ocurrir en pacientes cuyo virus contiene una o ambas sustituciones de aminoácidos. Emtricitabina: los aislados resistentes a la emtricitabina (M184V/I) manifestaron una resistencia cruzada con respecto a lamivudina y zalcitabina, pero conservaron la susceptibilidad in vitro en cuanto a didanosina, estavudina, tenofovir, zidovudina y los inhibidores no nucleósidos de la transcriptasa inversa (delavirdina, efavirenz y nevirapina). Los aislados de pacientes que han recibido un tratamiento intenso que contienen la sustitución de aminoácido M184V/I en el contexto de otras sustituciones de inhibidores nucleósidos de la transcriptasa inversa asociadas a la resistencia pueden conservar la susceptibilidad a tenofovir. Los aislados de VIH-1 que contienen la sustitución K65R, seleccionada in vivo por medio de abacavir, didanosina, tenofovir y zalcitabina, demostraron una susceptibilidad reducida con respecto a la inhibición mediante emtricitabina. Los virus con mutaciones que confieren una susceptibilidad reducida a estavudina y zidovudina (M41L, D67N, K70R, L210W, T215Y/F, K219Q/E) o a didanosina (L74V) permanecieron sensibles a la emtricitabina. El VIH-1 que contiene la sustitución K103N asociada a la resistencia a los inhibidores no nucleósidos de la transcriptasa inversa se mostró susceptible a la emtricitabina. Tenofovir disoproxil fumarato: los aislados de VIH-1 de pacientes (N=20) cuyo VIH-1 manifestó una media de 3 sustituciones de aminoácidos de transcriptasa inversa asociadas a zidovudina (M41L, D67N, K70R, L210W, T215Y/F o K219Q/E/N) indicaron una disminución de 3,1 veces en la susceptibilidad a tenofovir. El VIH-1 resistente a los nucleósidos múltiples con una mutación de inserción doble T69S en la transcriptasa inversa indicó una reducción en la susceptibilidad a tenofovir. Farmacología clínica: farmacocinética en adultos: TRUVADA: un comprimido de TRUVADA resultó bioequivalente a una cápsula de EMTRIVA (200mg) más un comprimido de VIREAD (300mg) tras la administración de una dosis simple a pacientes sanos en ayunas (N=39). Emtricitabina: las propiedades farmacocinéticas de la emtricitabina se resumen en la tabla 1. Luego de la administración oral de EMTRIVA, la emtricitabina se absorbe con rapidez con concentraciones pico de plasma a la hora o a las 2 horas posteriores a la dosis. La ligazón in vitro de emtricitabina con las proteínas del plasma humano es < 4% e independiente de la concentración en el rango de 0,02-200mg/ml. Tras la administración de emtricitabina radiomarcada, un 86% aproximadamente se recupera en la orina y un 13% se recupera en forma de metabolitos. Los metabolitos de la emtricitabina incluyen 3'-sulfóxido diastereómeros y su conjugado de ácido glucurónico. La emtricitabina se elimina mediante una combinación de filtración glomerular y secreción tubular activa. Luego de una dosis oral simple de EMTRIVA, la semivida de la emtricitabina en plasma es de 10 horas aproximadamente. Tenofovir disoproxil fumarato: las propiedades farmacocinéticas del tenofovir disoproxil fumarato se resumen en la tabla 1. Tras la administración oral de VIREAD, las concentraciones máximas de tenofovir en suero se lograron después de 1,0 ± 0,4 hora. La ligazón in vitro del tenofovir con las proteínas del plasma humano es de < 0,7% e independiente de la concentración en el rango de 0,01-25mg/ml. Aproximadamente un 70-80% de la dosis intravenosa de tenofovir se recupera en forma de droga no modificada en la orina. El tenofovir se elimina mediante una combinación de filtración glomerular y secreción tubular activa. Luego de una dosis oral simple de VIREAD, la semivida de eliminación terminal del tenofovir es de 17 horas aproximadamente.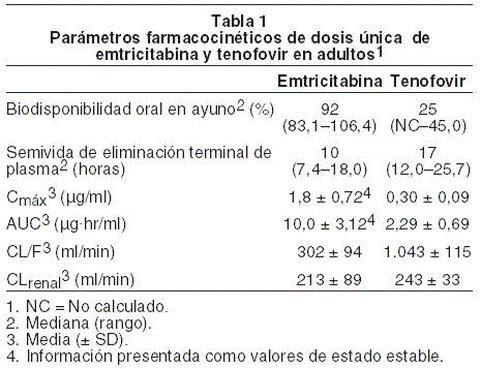
Efectos de la alimentación en la absorción oral: TRUVADA puede administrarse con o sin ingesta de alimentos. La administración de TRUVADA luego de una comida con alto contenido de grasas (784 kcal; 49 gramos de grasa) o de una comida liviana (373 kcal; 8 gramos de grasa) retrasó el tiempo de Cmáx de tenofovir en aproximadamente 0,75 hora. Los aumentos medios en AUC y Cmáx de tenofovir fueron aproximadamente de 35% y 15%, al ser administrados con una comida con alto contenido de grasas o con una comida liviana, en comparación con la administración en estado de ayuno. En estudios previos de seguridad y eficacia, se administró VIREAD (tenofovir) con ingesta de alimentos. Las exposiciones sistémicas de la emtricitabina (AUC y Cmáx) no se vieron afectadas cuando se administró TRUVADA ya sea con una comida con alto contenido de grasas o bien con una comida liviana. Poblaciones especiales: raza: emtricitabina: no se han identificado diferencias farmacocinéticas debidas a la raza tras la administración de emtricitavina. Tenofovir disoproxil fumarato: se dispuso de un número insuficiente de grupos raciales y étnicos que no fueran el caucásico para determinar de manera adecuada las potenciales diferencias farmacocinéticas entre estas poblaciones tras la administración de VIREAD. Género: emtricitabina y tenofovir disoproxil fumarato: la farmacocinética de la emtricitabina y del tenofovir es similar en pacientes de sexo masculino y femenino. Pacientes pediátricos y geriátricos: la farmacocinética de la emtricitabina y del tenofovir no se evaluó por completo en niños (menores de 18 años) o personas de la tercera edad (mayores de 65 años) (ver Precauciones, Uso pediátrico, Uso geriátrico). Pacientes con función renal insuficiente: la farmacocinética de emtricitabina y de tenofovir se ve alterada en pacientes con insuficiencia renal (ver Advertencias, Insuficiencia renal). En los pacientes con una depuración de creatinina menor a 50ml/min, la Cmáx y AUC0-inf de emtricitabina y de tenofovir se vieron incrementadas. Se recomienda que el intervalo de dosificación de TRUVADA se modifique en los pacientes con una depuración de creatinina de 30-49ml/min. TRUVADA no debería utilizarse en pacientes con una depuración de creatinina menor a 30ml/min y en pacientes con una enfermedad renal en su fase final que requiera diálisis (ver Advertencias, Insuficiencia renal). Pacientes con insuficiencia hepática: la farmacocinética de tenofovir luego de una dosis de 300mg de VIREAD se ha estudiado en pacientes no infectados con el VIH con una insuficiencia hepática de moderada a grave. No se produjeron alteraciones sustanciales en la farmacocinética del tenofovir en los pacientes con una insuficiencia hepática en comparación con los pacientes sin insuficiencia alguna. La farmacocinética de TRUVADA o de emtricitabina no se ha estudiado en pacientes con insuficiencia hepática. No obstante, la emtricitabina no es metabolizada de manera significativa por las enzimas del hígado, por lo tanto el impacto de la insuficiencia del hígado debería limitarse. Embarazo: (ver Precauciones, Embarazo). Madres lactantes: (ver Precauciones, Madres lactantes). Interacción de drogas: (ver Precauciones, Interacción de drogas). TRUVADA: no se han realizado estudios de interacción de drogas utilizando comprimidos de TRUVADA. Emtricitabina y tenofovir disoproxil fumarato: la farmacocinética en estado estable de emtricitabina y tenofovir disoproxil fumarato no se vio afectada cuando se administró emtricitabina y tenofovir disoproxil fumarato juntos al contrario de cuando se administró cada agente por separado. Los estudios in vitro y clínicos de interacción droga-droga de farmacocinética han demostrado que el potencial para las interacciones mediadas con enzimas citocromo P-450 que incluyen a la emtricitabina y al tenofovir y a otros productos medicinales es bajo. Emtricitabina y tenofovir son excretados primariamente por los riñones mediante una combinación de filtración glomerular y secreción tubular activa. No se observaron interacciones droga-droga debido a la competencia por la excreción renal. No obstante, la administración conjunta de TRUVADA y drogas que se eliminan mediante secreción tubular activa puede aumentar las concentraciones de emtricitabina, tenofovir y/o la droga administrada de manera conjunta. Las drogas que disminuyen la función renal pueden aumentar las concentraciones de emtricitabina y/o de tenofovir. No se observaron interacciones de drogas clínicamente significativas entre emtricitabina y famciclovir, indinavir, estavudina y tenofovir disoproxil fumarato. Asimismo, no se observaron interacciones de drogas clínicamente significativas entre tenofovir disoproxil fumarato y abacavir, adefovir dipivoxil, ribavirin, efavirenz, emtricitabina, indinavir, lamivudina, lopinavir/ritonavir, metadona y los anticonceptivos orales en estudios llevados a cabo en voluntarios sanos. Luego de dosis múltiples a personas con VIH negativo que reciben una terapia crónica de mantenimiento con metadona o anticonceptivos orales, o dosis simples de ribavarin, la farmacocinética del tenofovir en estado estable fue similar a la observada en estudios previos, lo que indica la falta de interacciones de drogas clínicamente significativas entre estos agentes y VIREAD. La administración conjunta de tenofovir disoproxil fumarato y didanosina produce modificaciones en la farmacocinética de didanosina que pueden tener relevancia clínica. La administración concomitante de tenofovir disoproxil fumarato y comprimidos tamponados o cápsulas con capa entérica de didanosina aumenta de manera significativa la Cmáx y AUC de didanosina. Cuando las cápsulas con capa entérica de didanosina de 250mg se administraron junto con tenofovir disoproxil fumarato, las exposiciones sistémicas de didanosina resultaron similares a las observadas en las cápsulas con capa entérica de 400mg en forma independiente en ayunas. Se desconoce el mecanismo de esta interacción.
Indicaciones.
TRUVADA se recomienda en combinación con otros agentes antirretrovirales (tales como inhibidores no nucleósidos de la transcriptasa inversa o inhibidores de la proteasa) para el tratamiento de la infección por VIH-1 en adultos. Los estudios de seguridad y eficacia con respecto al uso de los comprimidos de TRUVADA o de EMTRIVA y VIREAD en combinación se hallan en curso. EMTRIVA y VIREAD han sido estudiados como parte de regímenes de drogas múltiples y se ha comprobado que son seguros y eficaces. En el estudio clínico 303, EMTRIVA y la lamivudina (3TC) demostraron patrones de eficacia, seguridad y resistencia comparables como parte de regímenes de drogas múltiples. Esta información y la obtenida en el estudio 903, en el que la lamivudina y el tenofovir se utilizaron en combinación, respalda el uso de los comprimidos de TRUVADA para el tratamiento de la infección por VIH-1 en adultos que no han recibido tratamiento previo (ver Descripción de estudios clínicos y Reacciones adversas). En los pacientes que han recibido tratamiento previo, el uso de TRUVADA debe ser guiado mediante las pruebas de laboratorio y el historial de tratamiento (ver Microbiología). Información adicional importante en cuanto al uso de TRUVADA para el tratamiento de la infección por VIH-1: no existen resultados de estudios que demuestren el efecto de TRUVADA en el avance clínico del VIH-1. No se recomienda el uso de TRUVADA como componente de un régimen de nucleósido triple. Descripción de estudios clínicos: con respecto a los estudios de seguridad y eficacia al utilizar EMTRIVA o VIREAD en combinación con otros agentes antirretrovirales, consultar también la información de prescripción de estos productos. Los estudios de seguridad y eficacia al utilizar los comprimidos de TRUVADA o EMTRIVA y VIREAD en combinación se hallan en curso. EMTRIVA: estudio 303: EMTRIVA QD + terapia de antecedentes estables (SBT) en comparación con lamivudina BID + SBT. El estudio 303 consistió en un estudio de 48 semanas, abierto, multicéntrico y controlado con activos, que comparó a EMTRIVA (200mg QD) con la lamivudina, en combinación con la estavudina o la zidovudina y un inhibidor de la proteasa o un inhibidor no nucleósido de la transcriptasa inversa en 440 pacientes que seguían un régimen de drogas triple antirretroviral que contenía lamivudina durante al menos 12 semanas con anterioridad al inicio del estudio y que tenían un ARN del VIH-1 de < 400 copias/ml. Los pacientes se seleccionaron al azar (1:2) para continuar la terapia con lamivudina (150mg BID) o para cambiar a EMTRIVA (200mg QD). Todos los pacientes se mantuvieron en su régimen estable de antecedentes. Los pacientes tenían una edad media de 42 años (en un rango de 22-80), 86% era de sexo masculino, 64% caucásicos, 21% afroamericanos y 13% hispánicos. Los pacientes tenían un recuento CD4 medio de línea basal de células de 527 células/mm3 (en un rango de 37-1909) y un ARN medio de VIH de línea basal de plasma de 1,7 log10 copias/ml (en un rango de 1,7-4,0). La duración media de la terapia antirretroviral previa fue de 27,6 meses. El aumento medio de la línea basal en el recuento CD4 de células fue de 29 células/mm3 para el grupo EMTRIVA y de 61 células/mm3 para el grupo lamivudina. Durante las 48 semanas, en el grupo EMTRIVA 2 pacientes (0,7%) experimentaron un evento CDC Clase C nuevo, en comparación con los 2 pacientes (1,4%) del grupo lamivudina. VIREAD: estudio 903: VIREAD + lamivudina + efavirenz en comparación con estavudina + lamivudina + efavirenz. Se informaron los datos correspondientes a las 144 semanas del estudio 903, un estudio doble-ciego, multicéntrico y controlado con activos, que comparó a VIREAD (300mg QD), administrado en combinación con lamivudina y efavirenz, con estavudina, lamivudina y efavirenz en 600 pacientes que no habían recibido antirretrovirales previamente. Los pacientes tenían una edad media de 36 años (en un rango de 18-64), 74% era de sexo masculino, 64% caucásicos y 20% de color. El recuento CD4 medio de células de línea de base fue de 279 células/mm3 (en un rango de 3-956) y el ARN medio de VIH-1 de plasma de línea de base fue de 77.600 copias/ml (en un rango de 417-5.130.000). Los pacientes se estratificaron mediante el ARN de línea de base de VIH-1 y el recuento CD4. El cuarenta y tres por ciento de los pacientes poseía cargas virales de línea basal de > 100.000 copias/ml y el 39% poseía recuentos CD4 de células de < 200 células/mm3. El logro de concentraciones de ARN del VIH-1 en plasma inferiores a 400 copias/ml en la semana 144 fue similar entre los dos grupos de tratamiento para la población estratificada en línea de base sobre la base de la concentración de ARN del VIH-1 ( > o ≤100.000 copias/ml) y el recuento CD4 de células ( < o ≥200 células/mm3). Durante las 144 semanas de terapia, 62% y 58% de los pacientes en los grupos VIREAD y estavudina respectivamente lograron y mantuvieron < 50 copias/ml confirmadas de ARN del VIH-1. El aumento medio de la línea de base en el recuento CD4 de células fue de 263 células/mm3 para el grupo VIREAD y de 283 células/mm3 para el grupo estavudina. Durante las 144 semanas, once pacientes en el grupo VIREAD y nueve pacientes en el grupo estavudina experimentaron un nuevo evento CDC Clase C. Los análisis genotípicos de pacientes con fracasos virológicos señalaron que el desarrollo de mutaciones asociadas a efavirenz y a lamivudina ocurre con más frecuencia y sin diferencia alguna entre los grupos de tratamiento. La mutación K65R se dio en 8 pacientes en el grupo VIREAD y en 2 pacientes en el grupo estavudina. De los 8 pacientes que desarrollaron K65R en el grupo VIREAD durante las 144 semanas, 7 de estas ocurrieron en las primeras 48 semanas de tratamiento y una en la semana 96. En este estudio no se identificaron otras mutaciones que tuvieran como consecuencia la resistencia a VIREAD.
Dosificación.
La dosis de TRUVADA consiste en 1 comprimido (que contiene 200mg de emtricitabina y 300mg de tenofovir disoproxil fumarato) administrado una vez por día en forma oral con ingesta de alimentos o sin ella. Ajuste de dosis para insuficiencia renal: se produjo un aumento significativo en las exposiciones a drogas cuando se administró EMTRIVA o VIREAD a pacientes con una insuficiencia renal de moderada a severa. Por lo tanto, el intervalo de dosis de TRUVADA debería ajustarse en pacientes con una línea de base de depuración de creatinina de 30-49ml/min utilizando las recomendaciones incluidas en la tabla 2. La seguridad y la eficacia de estas recomendaciones concernientes al ajuste del intervalo de la dosis no se han evaluado a nivel clínico; por consiguiente, la respuesta clínica al tratamiento y la función renal deben ser controladas rigurosamente en estos pacientes.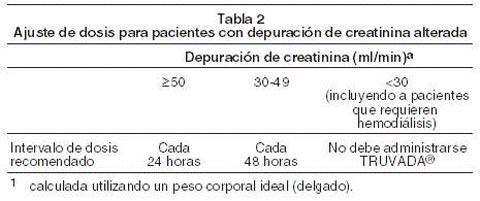
Contraindicaciones.
TRUVADA no se recomienda a pacientes con una hipersensibilidad a cualquiera de los componentes del producto que se haya manifestado previamente.
Reacciones adversas.
Ensayos clínicos: TRUVADA: los estudios de seguridad y eficacia al utilizar los comprimidos de TRUVADA o al utilizar EMTRIVA y VIREAD en combinación se hallan en curso. Doscientos ochenta y tres pacientes infectados con VIH-1 han recibido una terapia combinada de EMTRIVA y VIREAD con un inhibidor no nucleósido de la transcriptasa inversa o un inhibidor de la proteasa durante entre 24 y 48 semanas en estudios clínicos en curso. Sobre la base de estos datos limitados, no se identificaron nuevos patrones de eventos adversos y no se observó aumento alguno en la frecuencia de las toxicidades establecidas. Para obtener información adicional sobre la seguridad en cuanto a EMTRIVA o VIREAD en combinación con otros agentes antirretrovirales, consultar también la información de prescripción de estos productos. EMTRIVA: los eventos adversos que ocurrieron en más del 5% de los pacientes que reciben EMTRIVA junto con otros agentes antirretrovirales en los ensayos clínicos incluyen: dolor abdominal, astenia, cefalea, diarrea, náuseas, vómitos, mareos y erupciones cutáneas (incluyendo sarpullidos, prurito, erupciones maculopapulares, urticaria, erupciones ampollosas, erupciones pustulares y reacciones alérgicas). Aproximadamente 1% de los pacientes interrumpió su participación en los estudios clínicos debido a estos eventos adversos. Entre otros eventos adversos observados se incluyen: dispepsia, artralgia, mialgia, sueños anormales, depresión, insomnio, neuropatía, neuritis periférica, parestesia, tos crónica y rinitis. Todos los eventos adversos se observaron con similar frecuencia en los grupos EMTRIVA y de tratamiento de control, a excepción de la decoloración cutánea, que se observó con mayor frecuencia en el grupo tratado con EMTRIVA. La decoloración cutánea, manifestada por la hiperpigmentación de las palmas de las manos y/o las plantas de los pies, por lo general fue leve y asintomática. El mecanismo y la relevancia clínica se desconocen. Se ha informado que, en 1-12% de los pacientes que reciben EMTRIVA, ocurrieron elevaciones de grado 3/4 de alanina aminotransferasa y aspartato aminotransferasa (5 x ULN), bilirrubina ( > 2,5 x ULN), quinasa creatina ( > 4 x ULN), disminución de neutrófilos ( < 750/mm3), amilasa pancreática ( > 2,0 x ULN), amilasa en suero ( > 2 x ULN), glucosa en suero ( < 40 o > 250mg/dl), lipasa en suero ( > 2,0 x ULN) y triglicéridos ( > 750mg/dl). VIREAD: los eventos adversos que ocurrieron en más del 5% de los pacientes que reciben VIREAD junto con otros agentes antirretrovirales en los ensayos clínicos incluyen: cefalea, náuseas, diarrea, vómitos, erupciones cutáneas (incluyendo sarpullidos, prurito, erupciones maculopapulares, urticaria, erupciones ampollosas y erupciones pustulares) y depresión. Menos de 1% de los pacientes interrumpió su participación en los estudios clínicos debido a eventos adversos gastrointestinales. Entre otros eventos adversos observados se incluyen: astenia, dolores, dolor abdominal, dolor de espalda, dolor de pecho, fiebre, flatulencias, mareos, dispepsia, anorexia, artralgia, lipodistrofia, insomnio, neuropatía periférica (incluyendo neuritis y neuropatía periférica), ansiedad, neumonía, sudoración, mialgia y pérdida de peso. Se ha informado que, en 2-12% de los pacientes que reciben VIREAD, ocurrieron elevaciones de grado 3/4 de alanina aminotransferasa y aspartato aminotransferasa ( > 5 x ULN), quinasa creatina ( > 4 x ULN), amilasa en suero ( > 2 x ULN), glucosa en orina ( > 3+), glucosa en suero ( > 250mg/dl) y triglicéridos en suero ( > 750mg/dl), hematuria ( > 100 RBC/HPF), colesterol total ( > 300mg/dl) y disminución de neutrófilos ( < 750/mm3). Experiencia posmarketing: EMTRIVA: no se han identificado eventos adicionales a ser incluidos en esta sección. VIREAD: además de los eventos adversos informados a partir de los ensayos clínicos, los siguientes eventos se han identificado durante el uso de VIREAD posterior a su aprobación. Debido a que se informaron en forma voluntaria sobre la base de una población de tamaño desconocido, no se pueden realizar estimaciones de frecuencia. Estos eventos han sido seleccionados para ser incluidos debido a una combinación de su seriedad, frecuencia o potencial conexión causal con VIREAD. Trastornos del sistema inmunitario: reacciones alérgicas. Trastornos en el metabolismo y la nutrición: hipofosfatemia, acidosis láctica. Desórdenes respiratorios, torácicos y mediastínicos: disnea. Trastornos gastrointestinales: dolor abdominal, aumento de amilasa, pancreatitis. Trastornos hepatobiliares: aumento de enzimas hepáticas, hepatitis. Trastornos renales y urinarios: insuficiencia renal, falla renal, falla renal aguda, síndrome de Fanconi, tubulopatía proximal, proteinuria, aumento de creatinina, necrosis tubular aguda, diabetes insípida nefrogénica, poliuria, nefritis intersticial (incluyendo casos agudos). Trastornos cutáneos y subcutáneos: rash. Trastornos musculoesqueléticos y del tejido conectivo: miopatía, osteomalacia (ambos asociados con tubulopatía renal proximal).
Precauciones.
Interacciones de drogas: tenofovir disoproxil fumarato: cuando se administró tenofovir disoproxil fumarato con didanosina, la Cmáx y AUC de didanosina administrada ya sea como formulación tamponada o con capa entérica aumentaron de manera significativa. Se desconoce el mecanismo de esta interacción. Las concentraciones de didanosina superiores podrían potenciar los eventos adversos asociados a didanosina, incluyendo pancreatitis y neuropatía. Se ha observado supresión del conteo de células CD4 en pacientes que han recibido tenofovir DF y didanosina en dosis de 400mg diarios. En los adultos con un peso superior a 60kg, la dosis de didanosina debería reducirse a 250mg cuando se administra en forma conjunta con TRUVADA. No existe información disponible para recomendar un ajuste de dosis de didanosina para los pacientes que pesan menos de 60kg. Al administrarse en forma conjunta, TRUVADA y VIDEX EC® pueden consumirse en ayunas o con una comida liviana ( < 400 kcal, 20% grasa). La administración conjunta de la formulación de un comprimido tamponado de didanosina y TRUVADA debe ocurrir en ayunas. La administración conjunta de TRUVADA y didanosina debe realizarse con precaución y los pacientes que reciben esta combinación deberían ser controlados rigurosamente en cuanto a eventos adversos asociados a la didanosina. El uso de didanosina debería interrumpirse en los pacientes que desarrollan eventos adversos asociados a la didanosina. Se ha demostrado que atazanavir y lopinavir/ritonavir aumentan las concentraciones de tenofovir. Se desconoce el mecanismo de esta interacción. Los pacientes que reciben atazanavir y lopinavir/ritonavir y TRUVADA deberían ser controlados en cuanto a eventos adversos asociados con TRUVADA. El uso de TRUVADA debería interrumpirse en los pacientes que desarrollan eventos adversos asociados a TRUVADA. El tenofovir reduce la AUC y la Cmáx de atazanavir. Cuando se administra de manera conjunta con TRUVADA, se recomienda la administración de atazanavir 300mg con ritonavir 100mg. Atazanavir sin ritonavir no debería administrarse junta con TRUVADA. Emtricitabina y tenofovir disoproxil fumarato: dado que emtricitabina y tenofovir son eliminados primariamente por los riñones, la administración conjunta de TRUVADA y drogas que reducen la función renal o compiten por la secreción tubular activa puede aumentar las concentraciones en suero de emtricitabina, tenofovir y/u otras drogas eliminadas por vía renal. Algunos ejemplos incluyen (pero no se limitan a) adefovir dipivoxil, cidofovir, aciclovir, valaciclovir, ganciclovir y valganciclovir. TRUVADA es una combinación de dosis fija de emtricitabina y tenofovir disoproxil fumarato. TRUVADA no debería administrarse en forma conjunta con EMTRIVA o VIREAD. Debido a las similitudes entre emtricitabina y lamivudina, TRUVADA no debería administrarse en forma conjunta con otras drogas que contengan lamivudina, incluyendo COMBIVIR®, EPIVIR®, EPIVIR-HBV®, EPZICOM® o TRIZIVIR®. Efectos óseos: tenofovir disoproxil fumarato: en el estudio 903, que duró 144 semanas, se observaron disminuciones de línea de base en la densidad mineral ósea de la columna lumbar y de la cadera en ambos grupos de estudio. En la semana 144, se obtuvo un porcentaje medio de disminución significativamente superior de línea de base en la densidad mineral ósea de la columna lumbar de pacientes que recibían VIREAD + lamivudina + efavirenz (-2,2% + 3,9) en comparación con pacientes que recibían estavudina + lamivudina + efavirenz (-1,0% + 4,6). Las modificaciones en la densidad mineral ósea de la cadera fueron similares entre los dos grupos de tratamiento (-2,8% + 3,5 en el grupo VIREAD contra -2,4% + 4,5 en el grupo estavudina). En ambos grupos, la mayor parte de la reducción en la densidad mineral ósea ocurrió durante las primeras 24-48 semanas de estudio y esta reducción se mantuvo a lo largo de las 144 semanas. Un veintiocho por ciento de los pacientes tratados con VIREAD contra un 21% de los pacientes tratados con estavudina perdieron al menos 5% de la densidad mineral ósea de la espina o 7% de la densidad mineral ósea de la cadera. Se informaron fracturas clínicamente relevantes (excluyendo las de dedos) en 4 pacientes del grupo VIREAD y en 6 pacientes del grupo estavudina. Además, se observaron aumentos significativos en los indicadores bioquímicos del metabolismo óseo (fosfatasa alcalina específicamente ósea en suero, osteocalcina en suero, telopéptido-C en suero y telopéptido-N urinario) en el grupo VIREAD en relación con el grupo estavudina, lo que sugiere un mayor recambio óseo. Los niveles de hormona paratiroidea en suero y de vitamina D 1,25 también fueron superiores en el grupo VIREAD con relación al grupo estavudina. Excepto por la fosfatasa alcalina específicamente ósea, estas modificaciones tuvieron como consecuencia valores que permanecieron dentro del rango normal. Se desconocen los efectos de los cambios relacionados con VIREAD en la densidad mineral ósea y en los indicadores bioquímicos en la salud ósea a largo plazo, así también el riesgo de fracturas futuras. Debe tenerse en cuenta el control óseo en los pacientes infectados con VIH que cuentan con un historial de fracturas óseas patológicas o corren el riesgo de padecer osteopenia. Si bien el efecto de la complementación con calcio y vitamina D no se ha estudiado, dicha complementación puede resultar beneficiosa para todos los pacientes. Si se sospecha la presencia de anormalidades óseas, debería obtenerse el asesoramiento apropiado. Redistribución de grasa: se ha observado una redistribución o acumulación de grasa corporal, incluyendo obesidad central, aumento de grasa dorsocervical ("joroba de búfalo"), pérdida de grasa periférica y facial, aumento del tamaño del busto y aspecto "cushingoide" en pacientes que reciben terapia antirretroviral. Actualmente, se desconoce el mecanismo y las consecuencias a largo plazo de estos eventos. Aún no se ha podido establecer una relación causal. Síndrome de reconstitución inmunológica: se ha observado un síndrome de reconstitución inmunológica en pacientes tratados con una terapia antirretroviral combinada, incluyendo VIREAD. Durante la fase inicial del tratamiento antirretroviral combinado, los pacientes cuyo sistema inmunitario responde pueden desarrollar una respuesta inflamatoria a infecciones oportunistas indolentes o residuales (tales como la infección Mycobacterium avium, el citomegalovirus, la neumonía Pneumocystis jirovecii o la tuberculosis), que pueden requerir una evaluación y un tratamiento más exhaustivos. Toxicología en animales: el tenofovir y el tenofovir disoproxil fumarato, administrados en estudios de toxicología a ratas, perros y monos en exposiciones (sobre la base de AUCs) superiores o equivalentes a 6 veces las observadas en humanos, causaron toxicidad ósea. En los monos, la toxicidad ósea se diagnosticó como osteomalacia. La osteomalacia observada en monos parecía ser reversible en virtud de la reducción de dosis o la interrupción del uso de tenofovir. En ratas y perros, la toxicidad ósea se manifestó en forma de densidad mineral ósea reducida. Se desconoce el mecanismo que subyace a la toxicidad ósea. Se detectaron evidencias de toxicidad renal en cuatro especies animales. Se observaron aumentos en la creatinina en suero, el nitrógeno ureico en sangre, la glucosuria, la proteinuria, la fosfaturia y/o la calciuria y disminuciones en el fosfato en suero en varios grados en estos animales. Dichas toxicidades se detectaron en exposiciones (sobre la base de las AUCs) de 2 a 20 veces superiores a las observadas en humanos. No se conoce la relación entre las anormalidades renales, en especial la fosfaturia, y la toxicidad ósea. Carcinogénesis, mutagénesis y deficiencia de la fertilidad: emtricitabina: los estudios de carcinogenicidad a largo plazo en ratas y ratones se hallan en curso. La emtricitabina no resultó genotóxica en los ensayos de prueba bacteriológica de reversión (prueba de Ames), de linfomas de ratones o de micronúcleos de ratones. La emtricitabina no afectó la fertilidad en exposiciones (AUC) 140 veces superiores aproximadamente en ratas macho o 60 veces superiores aproximadamente en ratones macho y hembra con respecto a las de seres humanos que recibían la dosis diaria recomendada de 200mg. La fertilidad fue normal en la cría de los ratones expuestos diariamente desde antes del nacimiento (en útero) hasta la madurez sexual a exposiciones diarias (concentraciones tiempo-plasma) aproximadamente 60 veces superiores a las de los seres humanos con la dosis diaria recomendada de 200mg. Tenofovir disoproxil fumarato: los estudios de carcinogenicidad oral a largo plazo del tenofovir disoproxil fumarato en ratones y ratas se llevaron a cabo en exposiciones de hasta aproximadamente 16 veces (en ratones) y 5 veces (en ratas) las observadas en seres humanos con respecto a la dosis terapéutica para la infección por VIH. En virtud de la dosis elevada, en los ratones hembra los adenomas hepáticos aumentaron con exposiciones 16 veces superiores a las de los seres humanos. En las ratas, el estudio resultó negativo en cuanto a los resultados carcinogénicos con exposiciones hasta 5 veces superiores a las observadas en los humanos según la dosis terapéutica. El tenofovir disoproxil fumarato resultó mutagénico en el ensayo in vitro con linfomas de ratones y negativo en una prueba bacteriológica in vitro de mutagenicidad (prueba de Ames). En un ensayo in vivo con micronúcleos de ratones, el tenofovir disoproxil fumarato resultó negativo al ser administrado a ratones macho