VALCOTE Comprimidos 500 mg
ABBOTT
Anticonvulsivo. Antiepiléptico.
Descripción.
El Divalproato de sodio es un compuesto estable combinado que incluye valproato de sodio y ácido valproico en una relación 1:1 molar y formado durante la neutralización parcial de ácido valproico con 0.5 equivalentes de hidróxido de sodio. El divalproato de sodio es designado químicamente como bis(2- propilpentanoato) ácido de sodio. El divalproato de sodio tiene un peso molecular de 310,41 y se presenta como un polvo blanco con un olor característico. Su fórmula empírica es C16H31NaO4. El divalproato de sodio comprimidos está pensado para administración oral. El divalproato de sodio comprimidos se presenta en tres formas equivalentes a 125 mg, 250 mg o 500 mg de ácido valproico. Ingredientes Inactivos: Polividona, almidón pregelatinizado, dióxido de silicio, talco, dióxido de titanio, ftalato de hipromelosa, monoglicéridos diacetilados, vainillina, colorante azul FD&C N°2 y colorante rojo D&C N°30.
Farmacología.
Farmacodinamia: El divalproato de sodio se disocia a ion valproato en el tracto gastrointestinal. Los mecanismos por los cuales el valproato ejerce sus efectos terapéuticos no se han establecido. Se ha sugerido que su actividad en la epilepsia está relacionada con las concentraciones crecientes en el cerebro del ácido gamma-aminobutírico (GABA). Farmacocinética - Absorción/Biodisponibilidad: Dosis orales equivalentes de productos de divalproato de sodio (Valcote) y cápsulas de ácido valproico (Depakene) liberan cantidades equivalentes de ión valproato sistémicamente. Aunque la tasa de absorción de ión valproato puede variar con la formulación administrada (líquida, sólida, o espolvoreado), las condiciones de uso (por ejemplo, en ayunas o postprandial) y el método de administración (es decir, ya sea que los contenidos de la cápsula sean espolvoreados sobre los alimentos o que se ingiera la cápsula intacta), estas diferencias deben ser de importancia clínica menor bajo condiciones de estado estable inalcanzadas con el uso crónico en el tratamiento de epilepsia. Sin embargo, es posible que las diferencias entre los diferentes productos de valproato en Tmáx y Cmáx pudieran ser importantes al inicio del tratamiento. Por ejemplo, en estudios de dosis única, el efecto de la alimentación tuvo una mayor influencia sobre la tasa de absorción del comprimido (incremento en Tmáx de 4 a 8 horas) que sobre la absorción de las cápsulas sprinkle (incremento en Tmáx de 3.3 a 4.8 horas.) A pesar que la tasa de absorción desde el tracto gastrointestinal y la fluctuación en las concentraciones plasmáticas de valproato varían con el regímen de dosificación y formulación, es improbable que la eficacia de valproato como un anticonvulsivante de uso crónico se vea afectada. La experiencia utilizando regímenes de dosificación de una vez al día a cuatro veces al día, así como también estudios realizados en modelos de primates epilépticos sometidos a una tasa constante de infusión, indican que la biodisponibilidad sistémica diaria total (extensión de la absorción) es la principal determinante del control de las convulsiones y que las diferencias en las razones de concentraciones plasmáticas máxima a mínima entre las formulaciones de valproato no son consecuentes desde un punto de vista clínico práctico. Se desconoce si la tasa de absorción influencia o no la eficacia de valproato como un agente antimaníaco o antimigrañoso. La administración conjunta de productos orales de valproato con alimentos y la sustitución entre las diferentes formulaciones de divalproato de sodio y ácido valproico no debe causar problemas clínicos en el manejo de pacientes con epilepsia (ver Dosificación). No obstante, cualquier cambio en el régimen de administración, o la adición o discontinuación de medicamentos concomitantes normalmente debe estar acompañada por un monitoreo cercano del status clínico y de las concentraciones plasmáticas de valproato. Farmacocinética - Distribución: Unión a Proteínas: La unión a proteínas plasmáticas de valproato es concentración dependiente y la fracción libre aumenta de aproximadamente 10% en 40 mcg/mL a 18,5% en 130 mcg/mL. La unión a proteínas de valproato se reduce en los ancianos, en pacientes con enfermedades hepáticas crónicas, en pacientes con daño renal, y en la presencia de otras drogas (e.j., aspirina). Inversamente, el valproato puede desplazar ciertas drogas de su unión a proteínas (e.j., fenitoína, carbamazepina, warfarina, y tolbutamida) (ver Interacciones para información más detallada sobre las interacciones farmacocinéticas del valproato con otras drogas). Distribución en el SNC: Las concentraciones de valproato en el líquido cefalorraquídeo (LCR) se aproximan a las concentraciones libres en el plasma (cerca del 10% de concentración total). Farmacocinética - Metabolismo: El valproato se metaboliza casi enteramente por el hígado. En pacientes adultos con monoterapia, 30 a 50% de una dosis administrada aparece en la orina como glucurónido conjugado. La b-oxidación mitocondrial es la otra vía metabólica importante, involucrando comúnmente a más del 40% de la dosis. Generalmente, menos de 15 a 20% de la dosis es eliminada por otros mecanismos oxidativos. Menos del 3% de una dosis administrada se excreta sin cambios en la orina. La relación entre la dosis y la concentración total de valproato es no lineal, la concentración no aumenta proporcionalmente con la dosis, sino que aumenta en una extensión menor debido a la unión a proteínas plasmáticas saturables. La cinética de la droga libre es lineal. Farmacocinética - Eliminación: El clearance plasmático medio y el volumen de distribución del valproato total son 0,56 L/hr/1.73 m2 y 11 L/1.73 m2, respectivamente. El clearance plasmático medio y el volumen de distribución para el valproato libre son de 4,6 L/hr/1.73 m2 y 92 L/1.73 m2. La vida media terminal media para la monoterapia con valproato varió entre 9 y 16 horas después de la administración de dosis orales de 250 a 1000 mg. Los estimados citados aplican principalmente a pacientes que no están recibiendo drogas que afectan los sistemas hepáticos de metabolización enzimática. Por ejemplo, los pacientes que están recibiendo drogas antiepilépticas inductoras de enzimas (carbamazepina, fenitoína, y fenobarbital) eliminarán más rápidamente el valproato. Debido a estos cambios en el clearance de valproato, se debe intensificar el monitoreo de las concentraciones del antiepiléptico cada vez que se introducen o retiran antiepilépticos concomitantes. Poblaciones especiales: Recién nacidos: Los niños dentro de los primeros dos meses de vida tienen una capacidad notoriamente reducida para eliminar valproato en comparación con los niños de mayor edad y los adultos. Esto es resultado de un menor clearance (quizás debido a un retardo en el desarrollo de la glucuronosiltransferasa y otros sistemas enzimáticos involucrados en la eliminación de valproato) así como también un mayor volumen de distribución (debido en parte a una menor unión a proteínas plasmáticas). Por ejemplo, en un estudio, la vida media en niños menores de diez días de edad varió de 10 a 67 horas en comparación con un rango de 7 a 13 horas en niños mayores a los dos meses. Niños: Los pacientes pediátricos (vale decir, entre los tres meses y diez años de edad) tienen clearances 50% más altos expresados sobre el peso (es decir, mL/min/kg) en comparación con adultos. Sobre los diez años de edad, los niños tienen parámetros farmacocinéticos que se aproximan a aquellos de los adultos. Ancianos: La capacidad de los pacientes ancianos (rango de edad: 68 a 89 años) para eliminar valproato ha mostrado ser reducida en comparación con los adultos más jóvenes (rango de edad: 22 a 26). El clearance intrínseco se reduce en 39%; la fracción libre de valproato se aumenta en 44%. Por consiguiente, la dosis inicial se debe reducir en los ancianos (ver Dosificación). Sexo: No se han observado diferencias en el clearance libre ajustado por área de superficie corporal entre sujetos masculinos y femeninos (4.8 ±0.17 y 4.7 ±0.07 L/hr por 1.73 m2, respectivamente). Origen étnico: No se han estudiado los efectos del origen étnico sobre la cinética de valproato. Enfermedad hepática: Ver Contraindicaciones y Advertencias. La enfermedad hepática deteriora la capacidad para eliminar valproato. En un estudio, el clearance de valproato libre se redujo en 50% en siete pacientes con cirrosis y en 16% en cuatro pacientes con hepatitis aguda, comparado con seis sujetos sanos. En ese estudio, la vida media de valproato se incrementó desde 12 a 18 horas. La enfermedad hepática también se asocia con concentraciones disminuidas de albúmina y mayores fracciones libres (incremento de 2 a 2.6 veces) de valproato. Por consiguiente, el monitoreo de las concentraciones totales puede llevar a error dado que las concentraciones libres pueden estar sustancialmente elevadas en pacientes con enfermedad hepática mientras que las concentraciones totales pueden parecer normales. Enfermedad renal: Se ha reportado una leve reducción (27%) en el clearance de valproato libre en pacientes con insuficiencia renal (clearence de creatinina < 10 mL/minuto); sin embargo, la hemodiálisis típicamente reduce las concentraciones de valproato en alrededor de 20%. Por lo tanto, no parece ser necesario efectuar ajustes de dosificación en pacientes con insuficiencia renal. La unión a proteínas en estos pacientes se reduce sustancialmente; por lo tanto, el monitoreo de las concentraciones totales puede llevar a error. Niveles Plasmáticos y Efecto Clínico: La relación entre concentración plasmática y respuesta clínica no está bien documentada. Un factor contribuyente es la unión a proteínas no lineal, concentración-dependiente de valproato que afecta el clearance de la droga. Por lo tanto, el monitoreo de valproato sérico total no puede entregar un índice confiable de las especies bioactivas de valproato. Por ejemplo, dado que la unión de valproato a las proteínas plasmáticas es concentración-dependiente, la fracción libre aumenta desde aproximadamente 10% en 40 mcg/mL a 18.5% en 130 mcg/mL. Fracciones libres más altas que lo esperado se presentan en los ancianos, en pacientes hiperlipidémicos y en pacientes con enfermedad hepática y renal. Epilepsia: El rango terapéutico en epilepsia comúnmente se considera ser de 50 a 100 mcg/mL de valproato total, aunque algunos pacientes pueden ser controlados con concentraciones plasmáticas más bajas o más elevadas. Manía: En estudios clínicos placebo-controlados en manía aguda, los pacientes fueron dosificados para una respuesta clínica con concentraciones plasmáticas mínimas entre 50 y 125 mcg/mL (ver Dosificación). Estudios Clínicos: Epilepsia: Crisis Parciales Complejas (CPC): La eficacia de divalproato de sodio para reducir la incidencia de crisis parciales complejas (CPC) que ocurren en forma aislada o en asociación con otros tipos de crisis se estableció en dos estudios controlados. En un estudio multiclínico, controlado con placebo, que utilizó un diseño aditivo (terapia adyuvante), 144 pacientes que continuaron experimentando ocho o más crisis parciales complejas (CPC) por ocho semanas durante un período de ocho semanas de monoterapia con dosis de ya sea carbamazepina o fenitoína suficientes para asegurar concentraciones plasmáticas dentro del "rango terapéutico" fueron randomizados para recibir, además de su droga antiepiléptica original (AED), ya sea divalproato de sodio o placebo. Los pacientes randomizados fueron seguidos por un total de 16 semanas. La tabla 5 presenta los hallazgos.
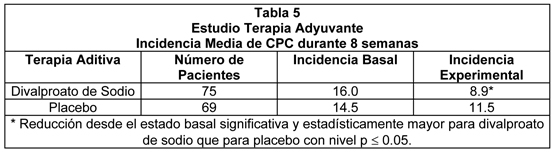
La Figura 1 presenta la proporción de pacientes (eje X) cuyo porcentaje de reducción desde el estado basal en las tasas de crisis parciales complejas fue al menos tan grande como la indicada en el eje Y en el estudio de terapia adyuvante. Un porcentaje de reducción positivo indica una mejoría (es decir, una disminución en la frecuencia de crisis), mientras que un porcentaje de reducción negativo indica un empeoramiento. Por lo tanto, en una imagen de este tipo, la curva para un tratamiento efectivo está desviada hacia la izquierda de la curva para placebo. Esta figura muestra que la proporción de pacientes que lograron algún nivel específico de mejoría fue consistentemente mayor para divalproato de sodio que para placebo. Por ejemplo, 45% de los pacientes tratados con divalproato de sodio tuvo una reducción ≥50% en la tasa de crisis parciales complejas comparado con 23% de los pacientes tratados con placebo.
El segundo estudio evaluó la capacidad de divalproato de sodio para reducir la incidencia de crisis parciales complejas (CPC) al ser administrado como droga antiepiléptica (AED) única. El estudio comparó la incidencia de CPC entre los pacientes randomizados ya sea a un brazo de tratamiento con dosis alta o uno con dosis baja. Los pacientes calificaban para ingresar a la fase comparativa randomizada de este estudio sólo si: 1) continuaban experimentando una o más CPC por cuatro semanas durante un período de 8 a 12 semanas de duración de monoterapia con dosis adecuadas de una AED (vale decir, fenitoína, carbamazepina, fenobarbital, o primidona); y 2) habían realizado una transición exitosa a través de un intervalo de dos semanas a divalproato de sodio. Los pacientes que ingresaron a la fase randomizada se les llevó a su dosis objetivo asignada, retirando gradualmente su AED concomitante, y seguidos por un intervalo de hasta 22 semanas. Sin embargo, menos del 50% de los pacientes randomizados completaron el estudio. En pacientes convertidos a monoterapia con divalproato de sodio, las concentraciones totales medias de valproato durante monoterapia fueron 71 y 123 mcg/mL, en los grupos de dosis baja y dosis alta, respectivamente. La tabla 6 presenta los hallazgos para todos los pacientes randomizados que tuvieron al menos una evaluación post-randomización.
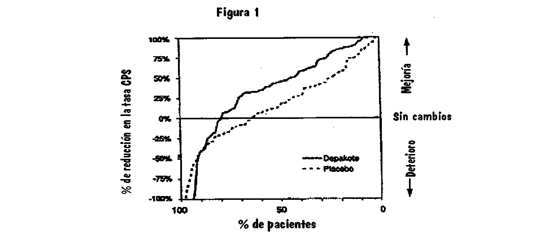
La Figura 2 presenta la proporción de pacientes (eje X) cuya reducción porcentual desde el estado basal en las tasas de crisis parciales complejas fue al menos tan elevada como la indicada en el eje Y en el estudio de monoterapia. Una reducción positiva del porcentaje indica una mejoría (vale decir, una disminución en la frecuencia de crisis), mientras que una reducción negativa del porcentaje indica empeoramiento. Por lo tanto, en una imagen de este tipo, la curva para un tratamiento más efectivo está desviada hacia la izquierda de la curva para un tratamiento menos efectivo. Esta figura muestra que la proporción de pacientes que lograron cualquier nivel dado de reducción fue consistentemente más alta para divalproato de sodio en dosis alta que para divalproato de sodio en dosis baja. Por ejemplo, cuando se cambia desde monoterapia con carbamazepina, fenitoína, fenobarbital o primidona a monoterapia con divalproato de sodio en dosis alta, 63% de los pacientes no experimentaron cambios o una reducción en las tasas de crisis parciales complejas comparado con 54% de los pacientes que recibieron divalproato de sodio en dosis baja.
Manía: La efectividad de divalproato de sodio en el tratamiento de la manía aguda se demostró en dos estudios de 3 semanas, controlados con placebo, grupo paralelo. Estudio 1: El primer estudio reclutó pacientes adultos que cumplían los criterios DSM-III-R para trastorno bipolar y que fueron hospitalizados por manía aguda. Además, tenían una historia de falla para responder a o intolerancia a tratamientos previos con carbonato de litio. El divalproato de sodio se inició en una dosis de 250 mg tres veces al día y se ajustó para alcanzar concentraciones séricas de valproato en un rango de 50 a 100 mcg/mL al día 7. Las dosis medias de divalproato de sodio para los que completaron este estudio fueron 1118, 1525, y 2402 mg/kg/día en los días 7, 14 y 21, respectivamente. Los pacientes se evaluaron de acuerdo al Young Mania Rating Scale (YMRS; rangos de puntaje desde 0 a 60), un Brief Psychiatric Rating Scale (BPRS-A) ampliado, y la Global Assessment Scale (GAS). Los análisis de los puntajes en el estado basal y el cambio desde el estado basal en el objetivo de la semana 3 (última observación llevada a cabo) fueron los siguientes:
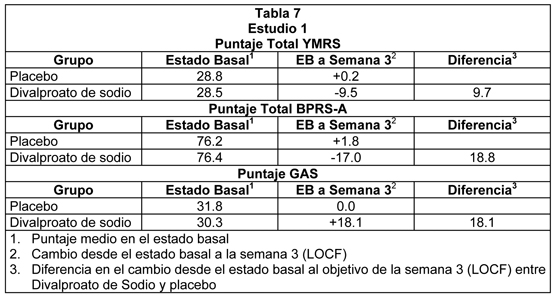
El divalproato de sodio fue estadística y significativamente superior a placebo en las tres mediciones de resultado. Estudio 2: El segundo estudio reclutó pacientes adultos que cumplieron los Criterios Diagnósticos de Investigación (Research Diagnostic Criteria) para trastorno maníaco y que fueron hospitalizados por manía aguda. Divalproato de sodio se inició en una dosis de 250 mg tres veces al día y se ajustó dentro de un rango de dosis de 750 a 2500 mg/día para alcanzar concentraciones séricas de valproato en un rango de 40 a 150 mcg/mL. Las dosis medias de divalproato de sodio para los pacientes que completaron este estudio fueron 1116, 1683 y 2006 mg/día en los días 7, 14 y 21, respectivamente. El estudio 2 también incluyó un grupo con litio para el cual las dosis de litio para quienes completaron el estudio fueron 1312, 1869, y 1984 mg/día en los días 7, 14, y 21, respectivamente. Los pacientes se evaluaron con la Manic Rating Scale (MRS; rangos de puntaje desde 11 a 63), y las medidas de objetivo primario fueron el puntaje MRS total, y los puntajes para 2 subescalas de la MRS, es decir, la Manic Syndrome Scale (MSS) y la Behavior and Ideation Scale (BIS). El análisis de los puntajes del estado basal y el cambio desde el estado basal en el objetivo de la semana 3 (última observación llevada a cabo) fueron los siguientes:
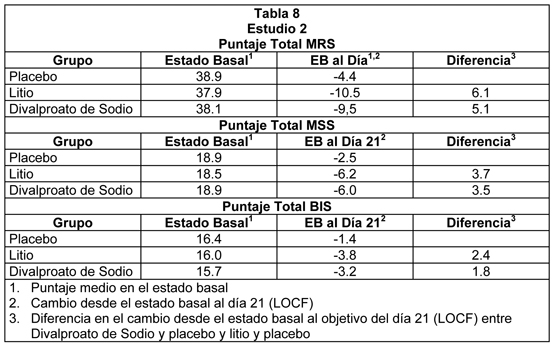
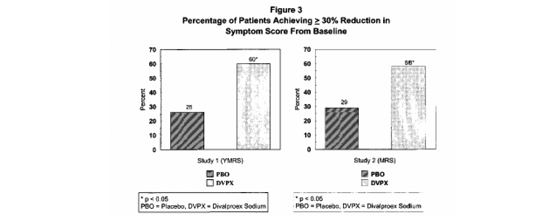
El divalproato de sodio fue estadística y significativamente superior a placebo en las tres mediciones de resultado. Un análisis exploratorio sobre los efectos de la edad y del género sobre el resultado no sugirió ninguna diferencia en la respuesta basados en la edad o género. Una comparación del porcentaje de pacientes que mostraron una reducción ≥30% en el puntaje de síntomas desde el estado basal en cada grupo de tratamiento, separados por estudio, se muestra en la Figura 3.
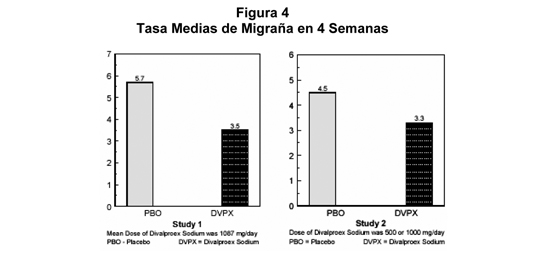
Migraña: Los resultados de dos estudios clínicos multicéntricos, randomizados, doble ciego, placebo-controlados establecieron la efectividad de divalproato de sodio en el tratamiento profiláctico de cefalea tipo migraña. Ambos estudios emplearon diseños esenciales idénticos y reclutaron pacientes con una historia de migraña con o sin aura (de al menos seis meses de duración) que experimentaron al menos dos cefaleas tipo migraña al mes durante los tres meses previos al reclutamiento. Los pacientes con cefaleas tipo cluster se excluyeron. Mujeres en edad fértil se excluyeron totalmente de un estudio, pero se permitieron en el otro si aceptaban usar un método anticonceptivo efectivo. En cada estudio después de un período basal de 4 semanas, simple ciego con placebo, los pacientes fueron randomizados, bajo condiciones de doble ciego, a divalproato de sodio o placebo para una fase de tratamiento de 12 semanas, que incluía un período de titulación de dosis de 4 semanas, seguido por un período de mantención de 8 semanas. El objetivo del tratamiento se evaluó en base a las tasas de cefalea tipo migraña en cuatro semanas, durante la fase de tratamiento. En el primer estudio, un total de 107 pacientes (24 hombres, 83 mujeres), en un rango etario desde 26 a 73 años fueron randomizados en una relación 2:1, a divalproato de sodio o placebo. Noventa pacientes completaron el período de mantención de 8 semanas. La titulación de dosis de la droga, usando comprimidos de 250 mg, fue individualizada a discreción del investigador. Los ajustes fueron guiados por los niveles séricos mínimos de valproato total, reales o supuestos para mantener el estudio ciego. En pacientes con divalproato de sodio, las dosis variaron desde 500 a 2500 mg al día. Las dosis sobre 500 mg fueron dadas en tres dosis divididas (tres veces al día). La dosis media durante la fase de tratamiento fue 1087 mg/día dando por resultado un nivel medio mínimo de valproato total de 72,5 mcg/mL, con un rango de 31 a 133 mcg/mL. La tasa media de cefalea tipo migraña en cuatro semanas durante la fase de tratamiento fue 5,7 en el grupo placebo comparado con 3.5 en el grupo de divalproato de sodio (ver Figura 4). Estas tasas fueron significativamente diferentes. En el segundo estudio, un total de 176 pacientes (19 hombres y 157 mujeres), en un rango etario desde 17 a 76 años, fueron randomizados equilibradamente a uno de tres grupos de dosis de divalproato de sodio (500, 1000, ó 1500 mg/día) o placebo. Los tratamientos fueron administrados en dos dosis divididas (dos veces al día). Ciento treinta y siete pacientes completaron el período de mantención de ocho semanas. La eficacia fue determinada por una comparación de la tasa de cefalea tipo migraña en cuatro semanas en el grupo combinado de 1000/1500 mg/día y el grupo placebo. La dosis inicial fue 250 mg diarios. El régimen se incrementó en 250 mg cada cuatro días (ocho días para el grupo 500 mg/día), hasta alcanzar la dosis randomizada. Los niveles medios mínimos de valproato total durante la fase de tratamiento fueron 39,6, 62,5, y 72,5 mcg/mL en los grupos de divalproato de sodio 500, 1000 y 1500 mg/día, respectivamente. Las tasas medias de cefalea tipo migraña en 4 semanas durante la fase de tratamiento, ajustados por diferencias en las tasas del estado basal, fueron 4,5 en el grupo placebo, comparado con 3,3; 3,0, y 3,3 en los grupos dee divalproato de sodio 500, 1000, y 1500 mg/día, respectivamente, basados en resultados de intención de tratar (ver Figura 4). Las tasas de cefalea tipo migraña en el grupo combinado de divalproato de sodio 1000/1500 mg fueron significativamente más bajas que en el grupo placebo.
Datos de Seguridad Pre-Clínica: Carcinogénesis, Mutagénesis y Deterioro de la Fertilidad: Carcinogénesis: El ácido valproico se administró a ratas Sprague Dawley y ratones ICR (HA/ICR) en dosis de 80 y 170 mg/kg/día (aproximadamente 10 a 50% de la dosis diaria humana máxima en una base de mg/m2) durante dos años. Aunque se observaron una variedad de neoplasmas en ambas especies, los principales hallazgos fueron un incremento estadísticamente significativo en la incidencia de fibrosarcomas subcutáneos en las ratas macho que recibieron dosis alta de ácido valproico y una tendencia estadísticamente significativa dosis-relacionada de adenomas pulmonares benignos en ratas macho que recibieron ácido valproico. Se desconoce la significancia de estos hallazgos para seres humanos. Mutagénesis: El valproato no fue mutagénico en un ensayo bacteriano in vitro (test de Ames), no produjo efectos letales dominantes en ratones, y no incrementó la frecuencia de aberración cromosómica en un estudio de citogenicidad in vivo en ratas. En un estudio de niños epilépticos que estaban recibiendo valproato se ha reportado una mayor frecuencia de intercambio de cromátidas hermanas (SCE= Sister Chromatic Exchange), pero esta asociación no se observó en otro estudio realizado en adultos. Existe alguna evidencia que las mayores frecuencias de SCE pueden estar asociadas con epilepsia. Se desconoce la significancia biológica de un incremento en la frecuencia de SCE. Deterioro de la Fertilidad: Los estudios de toxicidad crónica en ratas y perros jóvenes y adultos demostraron una espermatogénesis disminuida y atrofia testicular a dosis orales de 400 mg/kg/día o más en ratas (aproximadamente equivalente a o superior a la dosis diaria humana máxima en una base de mg/m2) y 150 mg/kg/día o más en perros (aproximadamente 1.4 veces la dosis diaria humana máxima o más en una base de mg/m2). Los estudios de fertilidad del segmento I en ratas han mostrado que dosis orales de hasta 350 mg/kg/día (aproximadamente igual a la dosis diaria humana máxima en una base de mg/m2) durante 60 días no tuvieron efecto sobre la fertilidad. Se desconoce el efecto de valproato sobre el desarrollo testicular y sobre la producción de espermios y la fertilidad en seres humanos.
Indicaciones.
Epilepsia: El divalproato de sodio comprimidos está indicado como monoterapia y terapia adyuvante para el tratamiento de pacientes con convulsiones parciales complejas que se presenten ya sea en forma aislada o en asociación con otros tipos de trastornos convulsivos. El divalproato de sodio en comprimidos también está indicado como terapia única y adyuvante en el tratamiento de crisis de ausencia simples y complejas, y como terapia adyuvante en pacientes con múltiples tipos de trastornos convulsivos que incluyen crisis de ausencia. La ausencia simple se define como una muy breve obnubilación sensorial o pérdida de la conciencia acompañada de ciertas descargas epilépticas generalizadas sin otros signos clínicos detectables. La ausencia compleja es el término utilizado cuando también están presentes otros signos. Manía: El divalproato de sodio en comprimidos está indicado para el tratamiento de los episodios maníacos asociados al trastorno bipolar. Un episodio maníaco es un período distinguible de humor anormal y persistentemente elevado, expansivo, e irritable. Los síntomas típicos de la manía incluyen discurso explosivo, hiperactividad motora, necesidad reducida de sueño, vuelo de ideas, grandiosidad, juicio pobre, agresividad, y posible hostilidad. La eficacia de divalproato de sodio fue establecida en estudios de tres semanas de duración en pacientes que cumplían con los criterios DSM-III-R para trastorno bipolar, que fueron hospitalizados por manía aguda (ver Estudios Clínicos). La seguridad y la eficacia del divalproato de sodio para el uso a largo plazo en manía, es decir, más de tres semanas, no se ha evaluado sistemáticamente en estudios clínicos controlados. Por lo tanto, los médicos que eligen utilizar el divalproato de sodio por períodos largos deben reevaluar continuamente la utilidad a largo plazo de la droga para cada paciente individual. Migraña: Los comprimidos de divalproato de sodio están indicados en la profilaxis de cefalea tipo migraña. No hay evidencia que el divalproato de sodio sea útil en el tratamiento agudo de la cefalea tipo migraña. Debido a que el ácido valproico puede ser un peligro para el feto, el divalproato de sodio se debe considerar para mujeres en edad fértil sólo después que este riesgo se ha discutido a fondo con la paciente y se ha sopesado contra el potencial beneficio del tratamiento (ver Advertencias - Uso en el Embarazo y Precauciones - Información para los Pacientes). Ver Advertencias con respecto a disfunción hepática fatal.
Dosificación.
General: Los comprimidos de Divalproato de Sodio se administran por vía oral y deben ser tragados enteros sin masticar. Epilepsia: Los comprimidos de divalproato de sodio están indicados como monoterapia y terapia adyuvante de crisis parciales complejas en pacientes adultos y pediátricos con una edad límite inferior de 10 años, y en crisis de ausencia simples y complejas en adultos y adolescentes. Puesto que la dosificación de Divalproato de Sodio se titula en forma ascendente, se pueden afectar las concentraciones de fenobarbital, carbamazepina, y/o fenitoína (ver Interacciones). Crisis Parciales Complejas (CPC): Para adultos y niños de 10 años de edad o mayores: Monoterapia (Terapia inicial): El divalproato de sodio no se ha estudiado en forma sistemática como terapia inicial. Los pacientes deben iniciar la terapia con una dosis de 10 a 15 mg/kg/día. La dosificación se debe incrementar en 5 a 10 mg/kg/semana para lograr una óptima respuesta clínica. Normalmente, la respuesta clínica óptima se logra a dosis diarias inferiores a 60 mg/kg/día. Si no se ha logrado una respuesta clínica satisfactoria, se deben medir niveles plasmáticos para determinar si se encuentran o no dentro del rango terapéutico generalmente aceptado (50 a 100 mcg/mL). No se pueden efectuar recomendaciones relacionadas con la seguridad de valproato al utilizarse a dosis superiores a 60 mg/kg/día. La probabilidad de trombocitopenia aumenta en forma significativa a concentraciones plasmáticas mínimas de valproato total superiores a 110 mcg/mL en mujeres y 135 mcg/mL en hombres. El beneficio de un mejor control de las crisis con las dosis más altas se debe contrapesar contra la posibilidad de una mayor incidencia de reacciones adversas. Conversión a Monoterapia: Los pacientes deben iniciar la terapia con 10 a 15 mg/kg/día. Posteriormente la dosificación se podrá incrementar en 5 a 10 mg/kg/semana para lograr la óptima respuesta clínica. Normalmente, la respuesta clínica óptima se logra a dosis diarias inferiores a 60 mg/kg/día. Si no se ha logrado una respuesta clínica satisfactoria, se deben medir niveles plasmáticos para determinar si están o no en el rango terapéutico generalmente aceptado (50 a 100 mcg/mL). No se pueden efectuar recomendaciones relacionadas con la seguridad de valproato para su uso a dosis superiores a 60 mg/kg/día. Normalmente se puede reducir la dosificación de la droga antiepiléptica (AED) concomitante en aproximadamente un 25% cada dos semanas. Esta reducción puede comenzar al inicio de la terapia con divalproato de sodio, o demorada en una a dos semanas si existe una preocupación que es probable que se presenten crisis con una menor dosificación. La velocidad y duración de la discontinuación de la droga antiepiléptica concomitante puede presentar una amplia variación, y los pacientes se deben monitorear cercanamente durante este período por una mayor frecuencia de las crisis. Terapia Adyuvante: El divalproato de sodio puede ser agregado al régimen del paciente a una dosificación de 10 a 15 mg/kg/día. La dosificación puede incrementarse por 5 a 10 mg/kg/semana hasta lograr una óptima respuesta clínica. Normalmente, la respuesta clínica óptima se logra a dosis diarias inferiores a 60 mg/kg/día. Si no se ha logrado la respuesta clínica óptima, se deben medir niveles plasmáticos para determinar si están o no en el rango terapéutico generalmente aceptado (50 a 100 mcg/mL). No se pueden efectuar recomendaciones en relación a la seguridad de valproato para su uso a dosis superiores a 60 mg/kg/día. Si la dosis diaria total excede de 250 mg, se debe administrar en dosis divididas. En un estudio de terapia adyuvante para crisis parciales complejas en el cual los pacientes estaban recibiendo ya sea carbamazepina o fenitoína en adición a divalproato de sodio, no se requirieron ajustes de dosificación de carbamazepina o fenitoína (ver Estudios Clínicos). Sin embargo, puesto que el valproato puede interactuar con estos o con otras drogas antiepilépticas administradas concomitantemente así como con otras drogas (ver Interacciones), se recomienda realizar determinaciones periódicas de las concentraciones plasmáticas de las drogas antiepilépticas concomitantes durante el curso temprano de la terapia (ver Interacciones). Crisis de Ausencia Simple y Compleja: La dosis inicial recomendada es de 15 mg/kg/día, aumentando a intervalos de una semana en 5 a 10 mg/kg/día hasta que se controlen las crisis o los efectos adversos impidan los futuros incrementos. La dosis máxima recomendada es de 60 mg/kg/día. Si la dosis diaria total excede de 250 mg, se debe administrar en dosis divididas. No se ha establecido una buena correlación entre la dosis diaria, las concentraciones plasmáticas y el efecto terapéutico. Sin embargo, las concentraciones plasmáticas terapéuticas de valproato para la mayoría de los pacientes con crisis de ausencia variarán desde 50 a 100 mcg/mL. Algunos pacientes pueden ser controlados con concentraciones plasmáticas más bajas o más altas (ver Farmacología). Dado que la dosificación de valproato de sodio se titula hacia arriba, se pueden ver afectadas las concentraciones sanguíneas de fenobarbital y/o fenitoína (ver Precauciones). Las drogas antiepilépticas no se deben discontinuar en forma abrupta en pacientes en los cuales la droga se administra para evitar crisis mayores debido a la fuerte posibilidad de precipitar estatus epilépticos con hipoxia concurrente y riesgo a la vida. Conversión de Acido Valproico a Divalproato de Sodio: En los pacientes que reciben previamente terapia con ácido valproico (Depakene), los productos de Divalproato de Sodio (Valcote) se deben iniciar en igual dosis diaria y horario. Después de que estabilice al paciente en un producto de divalproato de sodio, un horario de dosificación de dos o tres veces al día se puede elegir en el paciente seleccionado. Manía: La dosis inicial recomendada es de 750 mg diarios en dosis divididas. La dosis se debe aumentar lo más rápido posible para alcanzar la dosis terapéutica más baja que produce el efecto clínico deseado o el rango deseado de concentración plasmática. En estudios clínicos controlados con placebo en manía aguda, los pacientes fueron dosificados para una respuesta clínica con un nivel de concentración plasmática entre 50 y 125 mcg/mL. Las concentraciones máximas fueron alcanzadas generalmente dentro de los 14 días. La dosificación recomendada máxima es 60 mg/kg/día. No hay evidencia disponible de estudios controlados para guiar a un clínico en el manejo a largo plazo de un paciente que mejora durante el tratamiento con divalproato de sodio de un episodio de manía aguda. Mientras que generalmente se está de acuerdo que el tratamiento farmacológico es deseable más allá de una respuesta aguda en la manía, para el mantenimiento de la respuesta inicial y para la prevención de nuevos episodios maníacos, no hay datos sistemáticos obtenidos para apoyar las ventajas de divalproato de sodio en el tratamiento a largo plazo. Aunque no hay datos de la eficacia que se refieran específicamente a tratamiento antimaníaco a largo plazo con divalproato de sodio, la seguridad del divalproato de sodio en uso a largo plazo es apoyada por el registro de datos que involucran a aproximadamente 360 pacientes tratados con divalproato de sodio por más de tres meses. Migraña: La dosis de inicio recomendada es de 250 mg dos veces al día. Algunos pacientes se pueden beneficiar con dosis de hasta 1000 mg/día. En los estudios clínicos, no hay evidencia que dosis más altas condujeran a mayor eficacia. Recomendaciones Generales de Dosificación: Dosificación en Pacientes Ancianos: Debido a una reducción en el clearance del valproato no unido y posiblemente a una mayor sensibilidad a la somnolencia en ancianos, la dosis de inicio se debe reducir en estos pacientes. Las dosis se deben incrementar más lentamente y con un monitoreo regular de la ingesta de líquidos y alimentos, deshidratación, somnolencia y otros eventos adversos. Se debe considerar la reducción de la dosis o la discontinuación de valproato en pacientes con una menor ingesta de alimentos o líquidos y en pacientes con excesiva somnolencia. La dosis terapéutica última se debe lograr en base a la respuesta clínica (ver Advertencias). Eventos Adversos Relacionados con la Dosis: La frecuencia de efectos adversos (especialmente elevación de las enzimas hepáticas y trombocitopenia) puede estar relacionada con la dosis. La probabilidad de trombocitopenia parece aumentar significativamente a concentraciones de valproato total de ≥110 mcg/mL (mujeres) o ≥135 mcg/mL (hombres) (ver Precauciones). El beneficio de un mejor efecto terapéutico con dosis más altas se debe contrapesar contra la posibilidad de una mayor incidencia de reacciones adversas. Irritación Gastrointestinal: Los pacientes que experimentan irritación gastrointestinal pueden beneficiarse con la administración de la droga con las comidas o al aumentar lentamente la dosis desde un nivel inicial bajo.
Contraindicaciones.
El divalproato de sodio no se debe administrar a pacientes con enfermedad hepática o disfunción hepática significativa. El divalproato de sodio está contraindicado en pacientes con hipersensibilidad conocida a la droga. El divalproato de sodio está contraindicado en pacientes con trastorno conocido del ciclo de la urea (ver Advertencias).
Reacciones adversas.
Manía: La incidencia de eventos tratamiento-emergentes se ha comprobado basado en datos combinados a partir de dos estudios clínicos placebo controlados con divalproato de sodio en el tratamiento de los episodios maníacos asociados con trastorno bipolar. Los eventos adversos fueron generalmente leves o moderados en intensidad, pero algunas veces fueron lo suficientemente serios para interrumpir el tratamiento. En estudios clínicos, las tasas de terminación prematura debido a intolerancia no fueron estadísticamente diferentes entre placebo, divalproato de sodio, y carbonato de litio. Un total de 4%, 8% y 11% de pacientes discontinuaron la terapia debido a intolerancia en los grupos de placebo, divalproato de sodio, y carbonato de litio, respectivamente. La Tabla 1 resume aquellos eventos adversos reportados para los pacientes en estos estudios donde la tasa de incidencia en el grupo tratado con divalproato de sodio fue mayor que 5% y mayor que la incidencia de placebo, o donde la incidencia del grupo tratado con divalproato de sodio era estadística y significativamente mayor que el grupo placebo. El vómito fue el único evento que fue reportado por significativamente (p ≤0.05) más pacientes que recibían divalproato de sodio comparado con placebo.
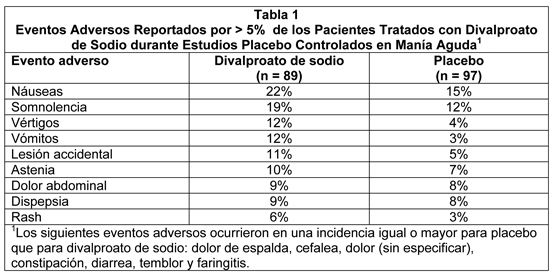
Los siguientes eventos adversos adicionales fueron reportados por más de 1% pero no más de 5% de los 89 pacientes tratados con divalproato de sodio en estudios clínicos controlados: Cuerpo como un Todo: Dolor de pecho, escalofríos, escalofríos y fiebre, quiste, fiebre, infección, dolor de cuello, rigidez del cuello. Sistema Cardiovascular: hipertensión, hipotensión, palpitaciones, hipotensión postural, taquicardia, vasodilatación. Sistema Digestivo: Anorexia, incontinencia fecal, flatulencia, gastroenteritis, glositis, absceso periodontal. Sistema Hemático y Linfático: Equimosis. Trastornos Metabólicos y Nutricionales: edema, edema periférico. Sistema Músculoesquelético: Artralgia, artrosis, calambres de la pierna, espasmo muscular. Sistema Nervioso: Sueños anormales, marcha anormal, agitación, ataxia, reacción catatónica, confusión, depresión, diplopía, disartria, alucinaciones, hipertonía, hipokinesia, insomnio, parestesia, reflejos aumentados, diskinesia tardía, pensamiento anormal, vértigo. Sistema Respiratorio: disnea, rinitis. Piel y Fanéreos: alopecia, lupus eritematoso discoide, piel seca, furunculosis, rash maculo papular, seborrea. Sentidos Especiales: ambliopía, conjuntivitis, sordera, ojos secos, dolor de oído, dolor de ojo, tinnitus. Sistema Urogenital: dismenorrea, disuria, incontinencia urinaria. Migraña: Basado en dos estudios clínicos controlados con placebo y su extensión a largo plazo, el Divalproato de Sodio fue generalmente bien tolerado con la mayoría de los eventos adversos clasificados como leves a moderados en severidad. De los 202 pacientes expuestos al divalproato de sodio en los estudios controlados con placebo, 17% discontinuaron por intolerancia. Esto se compara a una tasa de 5% para los 81 pac
ientes con placebo. Incluyendo la extensión del estudio a largo plazo, los eventos adversos reportados como razón primaria para discontinuación por ≥1% de 248 pacientes tratados con divalproato de sodio fueron alopecia (6%), náuseas y/o vómitos (5%), aumento de peso (2%), temblor (2%), somnolencia (1%), SGOT y/o SGPT elevados (1%), y depresión (1%). La tabla 2 incluye aquellos eventos adversos reportados para pacientes en los estudios placebo controlados donde la tasa de incidencia en el grupo tratado con divalproato de sodio fue mayor de 5% y fue mayor que para los pacientes con placebo.
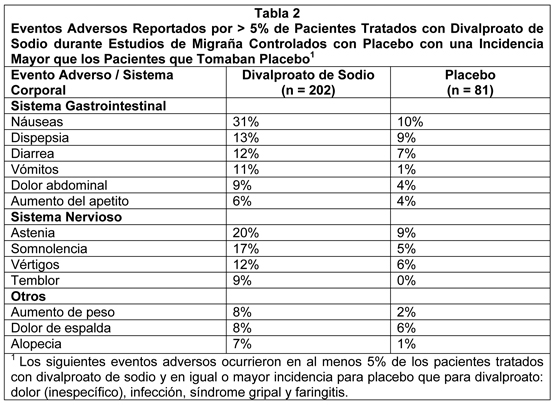
Los siguientes eventos adversos adicionales fueron reportados en más de 1% pero no más de 5% de los 202 pacientes tratados con divalproato de sodio en estudios clínicos controlados: Cuerpo como un Todo: Dolor en el pecho, escalofríos, edema facial, fiebre y malestar. Sistema Cardiovascular: Vasodilatación. Sistema Digestivo: Anorexia, constipación, boca seca, flatulencia, trastornos gastrointestinales (inespecíficos) y estomatitis. Sistema Hemático y Linfático: Equimosis. Trastornos Metabólicos y Nutricionales: Edema periférico, aumento del SGOT, aumento del SGPT. Sistema Músculoesquelético: Calambres de las piernas y mialgias. Sistema Nervioso: Sueños anormales, amnesia, confusión, depresión, labilidad emocional, insomnio, nerviosismo, parestesias, trastornos del habla, pensamiento anormal, vértigo. Sistema Respiratorio: Aumento de la tos, disnea, rinitis y sinusitis. Piel y Accesorios: Prurito, rash. Sentidos Especiales: Conjuntivitis, trastornos auditivos, perversión del gusto, tinnitus. Sistema Urogenital: Cistitis, metrorragia, hemorragia vaginal. Epilepsia: Crisis Parciales Complejas (CPC): Basado en un estudio controlado con placebo de terapia adyuvante para el tratamiento de crisis parciales complejas, el Divalproato de Sodio fue generalmente bien tolerado siendo la mayoría de los eventos adversos evaluados como leves a moderados en severidad. La intolerancia fue la razón principal para discontinuación en los pacientes tratados con Divalproato de Sodio (6%) comparado con 1% de los pacientes tratados con placebo. La Tabla 3 lista los eventos adversos tratamiento emergentes que fueron reportados por ≥5% de los pacientes tratados con Divalproato de Sodio y para los cuales la incidencia fue mayor que en el grupo placebo, en el estudio controlado con placebo de terapia adyuvante para el tratamiento de crisis parciales complejas. Puesto que los pacientes también fueron tratados con otras drogas antiepilépticas, no es posible, en la mayoría de los casos, determinar si los siguientes eventos adversos pueden ser adscritos a Divalproato de Sodio solo, o a la combinación de Divalproato de Sodio y otras drogas antiepilépticas.
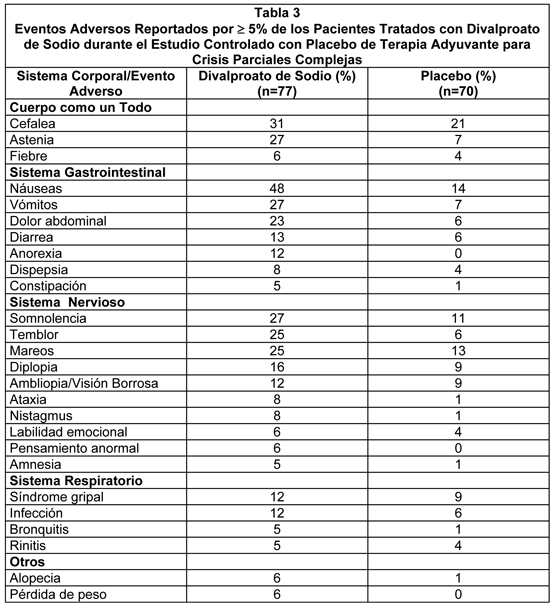
La Tabla 4 lista los eventos adversos tratamiento emergentes que fueron reportados por 5% de los pacientes en el grupo de divalproato de sodio de dosis alta, y para los cuales la incidencia fue superior que en el grupo de dosis baja, en un estudio controlado de monoterapia con divalproato de sodio en el tratamiento de crisis parciales complejas. Dado que los pacientes estaban siendo retirados de otra droga antiepiléptica durante la primera parte del estudio, no es posible, en muchos casos, determinar si los siguientes eventos adversos pueden ser adscritos a Divalproato de Sodio solo, o a la combinación de Divalproato de Sodio y otras drogas antiepilépticas.
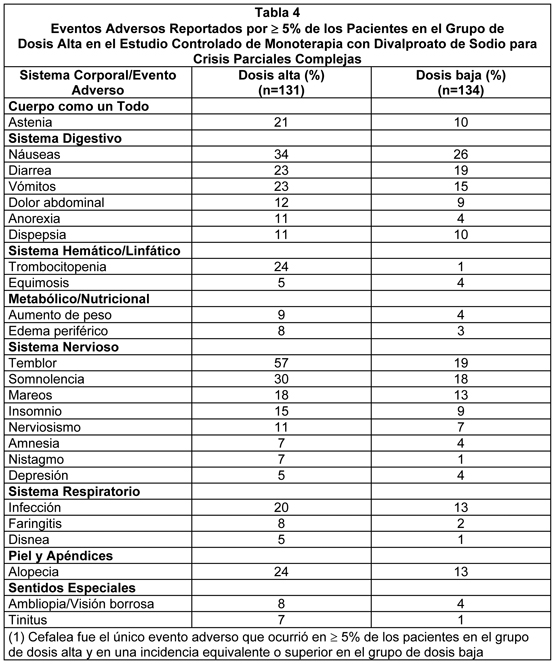
Los siguientes eventos adversos adicionales fueron reportados por más de 1% pero menos del 5% de los 358 pacientes tratados con divalproato de sodio en los estudios controlados de crisis parciales complejas: Cuerpo como un Todo: Lumbago, dolor en el pecho, malestar. Sistema Cardiovascular: Taquicardia, hipertensión, palpitaciones. Sistema Digestivo: Aumento del apetito, flatulencia, hematemesis, eructos, pancreatitis, absceso periodontal. Sistema Hemático y Linfático: Petequias. Trastornos Metabólicos y Nutricionales: elevación del SGOT, elevación del SGPT. Sistema Músculoesquelético: Mialgia, espasmo muscular, artralgia, calambres en las piernas, miastenia. Sistema Nervioso: Ansiedad, confusión, marcha anormal, parestesias, hipertonía, incoordinación, sueños anormales, trastornos de la personalidad. Sistema Respiratorio: Sinusitis, incremento en la tos, neumonía, epistaxis. Piel y Apéndices: Rash, prurito, piel seca. Sentidos Especiales: Perversión del gusto, visión anormal, sordera, otitis media. Sistema Urogenital: Incontinencia urinaria, vaginitis, dismenorrea, amenorrea, frecuencia de la micción. Otras poblaciones de pacientes: Los eventos adversos que se han reportado, se listan a continuación por sistema corporal con todas las presentaciones de dosis de valproato de los estudios en epilepsia, reportes espontáneos, y otras fuentes. Gastrointestinal: Los efectos adversos más comúnmente reportados al inicio de la terapia son náuseas, vómitos e indigestión. Estos efectos son generalmente transitorios y raramente requieren discontinuación de la terapia. Se han reportado diarrea, calambres abdominales y constipación. También se ha reportado tanto anorexia con cierta pérdida de peso como incremento en el apetito con aumento de peso. La administración de comprimidos con recubrimiento entérico de divalproato de sodio puede resultar en la reducción de los efectos colaterales gastrointestinales en algunos pacientes. Efectos sobre el SNC: Se han observado efectos sedantes en pacientes que reciben valproato solo, pero esto sucede con mayor frecuencia en los pacientes que reciben terapia combinada. La sedación generalmente remite con la reducción de otros medicamentos antiepilépticos. Temblor (puede ser dosis-relacionado), alucinaciones, ataxia, cefalea, nistagmo, diplopía, asterixis, "manchas ante los ojos", disartria, mareos, confusión, hiperestesia, vértigo, falta de coordinación y parkinsonismo han sido reportados con el uso de valproato. Casos raros de coma se han presentado en pacientes que reciben valproato solo o en conjunto con fenobarbital. En casos raros, se ha desarrollado encefalopatía con o sin fiebre poco después de la introducción de monoterapia con valproato sin evidencia de disfunción hepática o niveles inadecuadamente altos de valproato plasmático. Aunque se ha informado de recuperación después del retiro de la droga, han existido casos fatales en pacientes con encefalopatía hiperamonémica, particularmente en pacientes con trastornos del ciclo de la urea subyacentes (ver Advertencias - Trastornos del Ciclo de la Urea y Precauciones.) Numerosos reportes han indicado demencia reversible y pseudoatrofia reversible en asociación con la terapia con valproato. Dermatológicos: Caída transitoria del cabello, rash cutáneo, fotosensibilidad, prurito generalizado, eritema multiforme, y síndrome de Steven-Johnson. Se han reportado casos raros de necrólisis epidérmica tóxica incluyendo un caso fatal de un lactante de seis meses de edad que recibió valproato y numerosos otros medicamentos concomitantes. Un caso adicional de necrosis epidérmica tóxica que resultó en muerte fue reportado en un paciente de 35 años de edad con SIDA que recibía numerosos medicamentos concomitantes y con una historia de múltiples reacciones cutáneas a drogas. Se han reportado reacciones cutáneas serias con la administración concomitante de lamotrigina y valproato (ver Interacciones). Psiquiátrico: Alteración emocional, depresión, psicosis, agresión, hiperactividad, hostilidad y deterioro del comportamiento. Músculo-esquelético: Debilidad: Se han recibido reportes de disminución de la masa ósea, llevando potencialmente a osteoporosis y osteopenia, durante la terapia a largo plazo con medicamentos anticonvulsivantes, incluyendo valproato. Algunos estudios han indicado que el suplemento de calcio y de vitamina D puede ser beneficioso para los pacientes que están en terapia crónica con valproato. Hematológico: Trombocitopenia e inhibición de la fase secundaria de la agregación plaquetaria se pueden reflejar en tiempos de sangrado alterados, petequias, moretones, formación de hematomas, epistaxis y hemorragia franca (ver Precauciones - General e Interacciones). Linfocitosis relativa, macrocitosis, hipofibrinogenemia, leucopenia, eosinofilia, anemia incluyendo macrocítica con o sin deficiencia de folato, supresión de la médula ósea, pancitopenia, anemia aplástica, agranulocitosis y porfiria intermitente aguda. Hepática: Elevaciones menores de transaminasas (ej. SGOT y SGPT) y LDH son frecuentes y parecen ser dosis-relacionadas. Ocasionalmente, los resultados de las pruebas de laboratorio también pueden incluir elevaciones en la bilirrubina sérica y cambios anormales en otros tests de función hepática. Estos resultados pueden reflejar hepatotoxicidad potencialmente seria (ver Advertencias). Endocrino: Menstruaciones irregulares, amenorrea secundaria, agrandamiento de las mamas, galactorrea, e inflamación de la glándula parótida. Test anormales de función tiroidea (ver Precauciones - General). Han existido raros reportes espontáneos de enfermedad de ovario poliquístico. No se ha establecido una relación de causa y efecto. Pancreático: Pancreatitis aguda incluyendo casos fatales (ver Advertencias). Metabólicas: Hiperamonemia (ver Precauciones - General), hiponatremia, y secreción inadecuada de ADH. Han existido raros reportes de síndrome de Fanconi presentándose primordialmente en niños. Se han reportado menores concentraciones de carnitina aunque se desconoce su relevancia clínica. Se ha presentado hiperglicinemia (concentración elevada de la glicina plasmática) y ésta estuvo asociada con un resultado fatal en un paciente con hiperglicinemia no cetónica preexistente. Genitourinaria: Enuresis e infección al tracto urinario. Sentidos especiales: Se han reportado pérdida de la audición, ya sea reversible o irreversible; sin embargo, no se ha establecido una relación de causa y efecto. También se ha reportado dolor de oídos. Otros: Reacción alérgica, anafilaxis, edema de las extremidades, lupus eritematoso, dolor óseo, aumento de la tos, neumonía, otitis media, bradicardia, vasculitis cutánea, fiebre e hipotermia.
Precauciones.
Disfunción hepatica: Ver Contraindicaciones y Advertencias. Pancreatitis: Ver Advertencias. Hiperamonemia: Se ha reportado hiperamonemia en asociación con terapia con valproato y puede estar presente a pesar de pruebas de función hepática normales. En pacientes que desarrollan inexplicables episodios de letargo y vómitos o cambios en su estado mental, se debe considerar encefalopatía hiperamonémica y se debe medir el nivel de amonio. También se debe considerar hiperamonemia en pacientes que se presentan con hipotermia (ver Precauciones - Hipotermia). Si el amonio aumenta, la terapia de valproato se debe discontinuar. Deben ser iniciadas intervenciones apropiadas para el tratamiento de la hiperamonemia y tales pacientes someterse a investigación para trastornos del ciclo de la úrea subyacente (ver Contraindicaciones y Advertencias - Trastornos del Ciclo de la Urea y Precauciones - Hiperamonemia y Encefalopatía Asociada con el Uso Concomitante de Topiramato). Las elevaciones asintomáticas del amonio son más comunes y cuando se presentan requieren estrecho monitoreo de los niveles de amonio plasmáticos. Si la elevación persiste, se debe considerar la discontinuación de la terapia de valproato. Hiperamonemia y Encefalopatía Asociada al Uso Concomitante de Topiramato: La administración concomitante de topiramato y de ácido valproico se ha asociado con hiperamonemia con o sin encefalopatía en pacientes que han tolerado cualquiera de estas drogas por si solas. Los síntomas clínicos de encefalopatía hiperamonémica a menudo incluyen alteraciones agudas en el nivel de conciencia y/o función cognoscitiva con letargo o vómitos. La hipotermia también puede ser una manifestación de hiperamonemia (ver Precauciones - Hipotermia). En la mayoría de los casos, los síntomas y signos disminuyeron con la discontinuación de cualquiera de las drogas. Este evento adverso no es debido a una interacción farmacocinética. No se sabe si la monoterapia con topiramato se asocia con hiperamonemia. Pacientes con fallas innatas del metabolismo o reducción de la actividad mitocondrial hepática pueden estar en un riesgo mayor de hiperamonemia con o sin encefalopatía. Aunque no está estudiado, una interacción de topiramato y ácido valproico puede exacerbar defectos existentes o desenmascarar deficiencias en personas susceptibles (ver Contraindicaciones y Advertencias - Trastornos del Ciclo de la Urea y Precauciones - Hiperamonemia). Hipotermia: Hipotermia, definido como una caída no intencional en la temperatura del cuerpo a < 35°C (95°F), se ha reportado en asociación con la terapia con valproato tanto en conjunto con como en ausencia de hiperamonemia. Esta reacción adversa también puede ocurrir en pacientes que usan topiramato concomitante con valproato después del inicio del tratamiento con topiramato o después de aumentar la dosis diaria de topiramato (ver Interacciones de drogas - Topiramato). Se debe tener consideración para suprimir el valproato en pacientes que desarrollan hipotermia, el cual se puede manifestar por una variedad de anormalidades clínicas que incluyen letargia, confusión, coma y alteraciones significativas en otros sistemas de órganos importantes tales como los sistemas cardiovascular y respiratorio. El manejo y la evaluación clínica deben incluir examen de los niveles de amonio sanguíneo. General: Debido a reportes de trombocitopenia (ver Advertencias), inhibición de la fase secundaria de la agregación plaquetaria, y parámetros de coagulación anormal (por ejemplo, bajo fibrinógeno), se recomienda realizar recuentos de plaquetas, y pruebas de coagulación antes de iniciar la terapia y a intervalos periódicos durante la misma. Se recomienda que los pacientes que reciben divalproato de sodio sean monitoreados con recuento de plaquetas y parámetros de coagulación previa a cirugía electiva. En un estudio clínico de divalproato de sodio como monoterapia en pacientes con epilepsia, 34/126 pacientes (27%) que recibieron aproximadamente 50 mg/kg/día en promedio, presentaron al menos un valor de plaquetas ≤75 x 109/L. Aproximadamente la mitad de estos pacientes discontinuaron el tratamiento, con retorno de los recuentos de plaquetas a los valores normales. En los pacientes restantes, los recuentos de plaquetas se normalizaron mientras continuaban el tratamiento. En este estudio, la probabilidad de trombocitopenia pareció aumentar significativamente a concentraciones totales de valproato de ≥110 mcg/mL (mujeres) o ≥135 mcg/mL (hombres). La evidencia de hemorragia, equimosis o un trastorno de la hemostasis/coagulación es una indicación para reducción de la dosificación o discontinuación de la terapia. Dado que el divalproato de sodio puede interactuar con drogas administradas concomitantemente que son capaces de inducción enzimática, se recomienda la realización de determinaciones periódicas de concentración plasmática de valproato y de las drogas concomitantes durante el curso temprano de la terapia (ver Interacciones de drogas). El valproato se elimina parcialmente en la orina como un ceto-metabolito que puede llevar a una interpretación falsa del test de cetona urinaria. Han existido reportes de pruebas de función tiroidea alterada asociados con valproato. La significancia clínica de la misma es desconocida. Existen estudios in vitro que sugieren que el valproato estimula la replicación de los virus VIH y CMV bajo ciertas condiciones experimentales. La consecuencia clínica de esto, si es que existe alguna, es desconocida. Adicionalmente, la relevancia de estos hallazgos in vitro es incierta para los pacientes que están recibiendo terapia antiretroviral con máxima actividad supresora. A pesar de ello, estos datos se deben tener en mente al interpretar los resultados del monitoreo regular de la carga viral en pacientes VIH infectados que están recibiendo valproato o al realizar el seguimiento clínico de pacientes infectados por CMV. Reacción de Hipersensibilidad Multi-orgánica: Raramente se han reportado reacciones de hipersensibilidad multiorgánica en asociación temporal cercana después del inicio de la terapia con valproato en adultos y pacientes pediátricos (mediana de tiempo a la detección 21 días: rango 1 a 40). Aunque ha habido un número limitado de informes, muchos de estos casos produjeron hospitalización y se ha reportado al menos una muerte. Los signos y síntomas de este trastorno fueron diversos; sin embargo, los pacientes típicamente, aunque no exclusivamente, presentaron fiebre y rash asociado con compromiso de otro sistema, órgano. Otras manifestaciones asociadas pueden incluir linfoadenopatía, hepatitis, pruebas de función hepática alteradas, anormalidades hematológicas (ej., eosinofília, trombocitopenia, neutropenia), prurito, nefritis, oliguria, síndrome hepato renal, artralgia, y astenia. Debido a que el desorden es variable en su expresión, signos y síntomas de otros sistemas, órganos, no mencionados aquí, pueden ocurrir. Si se sospecha esta reacción, el valproato se debe discontinuar y comenzar un tratamiento alternativo. Aunque la existencia de sensibilidad cruzada con otras drogas que producen este síndrome no es clara, la experiencia entre las drogas asociadas con hipersensibilidad multi-orgánica indicaría ésta es una posibilidad. Información para los Pacientes: Se deberá informar a los pacientes y sus apoderados que el dolor abdominal, náuseas, vómitos y/o anorexia pueden ser síntomas de pancreatitis, y que, por consiguiente, requieren de una pronta evaluación médica adicional. Los pacientes y sus apoderados deben ser informados sobre los signos y síntomas asociados con la encefalopatía hiperamonémica (ver Precauciones - Hiperamonemia) y señalarles que deben informar al médico si cualquiera de estos síntomas aparece. Dado que el valproato de sodio puede producir depresión del SNC, especialmente al ser combinado con otro depresor del SNC (por ejemplo, el alcohol), se deberá recomendar a los pacientes que no realicen actividades riesgosas, tales como conducir un automóvil u operar máquinas peligrosas, hasta que se demuestre que no sufren de somnolencia con la droga. Puesto que el divalproato de sodio se ha asociado a ciertos tipos de defectos de nacimiento, a las pacientes mujeres en edad fértil que estén considerando tomar divalproato de sodio se les debe aconsejar sobre los riesgos asociados al uso del divalproato de sodio durante el embarazo (ver Advertencias). Uso Pediátrico: La experiencia ha indicado que los niños menores de dos años presentan un considerable mayor riesgo de desarrollar hepatotoxicidad fatal, especialmente aquellos con las condiciones antes mencionadas (ver Advertencias). Cuando se utiliza Divalproato de Sodio en este grupo de pacientes, se debe administrar con extremada cautela y como agente único. Los beneficios de la terapia se deben contrapesar contra los riesgos. Sobre los 2 años de edad, la experiencia en epilepsia ha indicado que la incidencia de hepatotoxicidad fatal se reduce considerablemente en grupos de pacientes progresivamente mayores. Los niños más jóvenes, especialmente aquellos que reciben drogas inductoras de las enzimas, requerirán dosis de mantenimiento más elevadas para obtener las concentraciones deseadas de ácido valproico libre y total. La variabilidad en la fracción libre limita la utilidad clínica del monitoreo de las concentraciones séricas totales de ácido valproico. La interpretación de las concentraciones de ácido valproico en niños debe incluir consideración de los factores que afectan el metabolismo hepático y la unión a proteínas. La seguridad y la eficacia del divalproato de sodio en el tratamiento de la manía aguda no han sido bien estudiadas en individuos menores de 18 años. La seguridad y la eficacia del divalproato de sodio para la profilaxis de migraña no han sido estudiadas en individuos menores de 16 años de edad. La toxicología básica y manifestaciones patológicas del divalproato de sodio en ratas recién nacidas (4 días de edad) y jóvenes (14 días de edad) son similares a aquellas observadas en ratas adultas jóvenes. Sin embargo, se han reportado hallazgos adicionales, incluyendo alteraciones renales en ratas jóvenes y alteraciones renales y displasia retinal en ratas recién nacidas. Estos hallazgos se presentaron a 240 mg/kg/día, una dosis que es aproximadamente equivalente a la dosis diaria máxima recomendada en seres humanos en una base mg/m2. No se observaron con 90 mg/kg, o 40% de la dosis diaria máxima en seres humanos en una base de mg/m2. Uso Geriátrico: No se han reclutado pacientes mayores de 65 años en estudios clínicos prospectivos, doble ciego de manía asociada con enfermedad bipolar. En un estudio de revisión de casos de 583 pacientes, 72 pacientes (12%) eran mayores de 65 años. Un porcentaje más alto de pacientes mayores de 65 años de edad reportó lesiones accidentales, infección, dolor, somnolencia y temblor. La discontinuación de valproato ocasionalmente se asoció con estos últimos dos eventos. No está claro si estos eventos indican un riesgo adicional o si resultan de enfermedades médicas preexistentes y el uso de medicamentos concomitantes entre estos pacientes. Un estudio de pacientes ancianos con demencia reveló somnolencia relacionada con la droga y discontinuación debido a somnolencia (ver Advertencias - Somnolencia en Ancianos). En estos pacientes se debe reducir la dosis de inicio, y se debe considerar reducción de la dosificación o discontinuación en pacientes con somnolencia excesiva (ver Dosificación). Se dispone de insuficiente información con relación a la seguridad y la eficacia del divalproato de sodio en profilaxis de migraña en pacientes mayores de 65 años.
Advertencias.
Hepatotoxicidad: Se ha presentado insuficiencia hepática con desenlace fatal en pacientes que han recibido ácido valproico. Estos incidentes generalmente se presentaron durante los primeros seis meses de tratamiento. La hepatotoxicidad seria o fatal puede estar precedida de síntomas inespecíficos tales como malestar, debilidad, letargo, edema facial, anorexia y vómitos. En pacientes con epilepsia, también se puede presentar una pérdida del control sobre las crisis. Los pacientes se deben monitorear de cerca por la aparición de estos síntomas. Las pruebas de función hepática se deben realizar antes de la terapia y durante intervalos frecuentes a continuación, especialmente durante los primeros seis meses. Sin embargo, los médicos no deben confiar únicamente en la bioquímica plasmática dado que estas pruebas pueden no ser anormales en todos los casos, sino que también deben considerar los resultados de una cuidadosa historia clínica y el examen físico. Se debe tener precaución al administrar productos con valproato de sodio a pacientes con una historia de enfermedad hepática previa. Los pacientes que reciben múltiples anticonvulsivantes, los niños, aquellos con trastornos metabólicos congénitos, con trastornos convulsivos severos acompañados por retardo mental y aquellos con enfermedad cerebral orgánica pueden estar en un riesgo especial. La experiencia ha indicado que los niños menores de dos años están en un riesgo considerablemente mayor de desarrollar hepatotoxicidad fatal, especialmente aquellos con las condiciones antes mencionadas. Cuando se usa Divalproato de Sodio en este grupo de pacientes, esto se debe hacer con extrema precaución y como agente único. Se deberán sopesar los beneficios de la terapia contra los posibles riesgos. Más allá de este grupo etario, la experiencia en epilepsia ha indicado que la incidencia de hepatotoxicidad fatal se reduce considerablemente en los grupos de pacientes progresivamente mayores. No se recomienda el uso de Divalproato de Sodio de liberación prolongada en niños para la profilaxis del dolor de cabeza tipo migraña (ver Precauciones - Uso en Pediatría). La droga se debe discontinuar inmediatamente en presencia de disfunción hepática significativa, sospechada o aparente. En algunos casos, la disfunción hepática ha progresado a pesar de la discontinuación de la droga. Pancreatitis: Se han reportado casos de pancreatitis con riesgo vital tanto en niños como en adultos que recibieron valproato. Algunos de los casos se han descrito como hemorrágicos con progresión rápida desde los síntomas iniciales a la muerte. Algunos casos se han presentado poco después del uso inicial, mientras que otros después de muchos años de uso. La tasa basada en los casos reportados excede lo esperado para la población general y han existido casos en los cuales la pancreatitis recurrió después de un nuevo tratamiento con valproato. En estudios clínicos, se han descrito dos casos de pancreatitis sin etiología alternativa en 2416 pacientes, representando 1044 pacientes-años de experiencia. Se debe alertar a los pacientes y sus cuidadores que el dolor abdominal, náuseas, vómitos y/o anorexia pueden ser síntomas de pancreatitis que requieren de rápida evaluación médica. Si se diagnostica pancreatitis, normalmente se debe discontinuar el valproato. El tratamiento alternativo para la condición médica subyacente se debe iniciar según indicación clínica (ver Advertencias). Trastornos del Ciclo de la Urea (TCU): La encefalopatía hiperamonémica, algunas veces fatal, se ha reportado después de la iniciación de la terapia con valproato en pacientes con trastornos del ciclo de la urea, un grupo de anormalidades genéticas poco comunes, particularmente deficiencia de la ornitina transcarbamilasa. Previo a la iniciación de la terapia con valproato, se debe considerar una evaluación de TCU en los siguientes pacientes: 1) aquellos con una historia de coma o encefalopatía inexplicable, encefalopatía asociada con carga proteica, encefalopatía relacionada con el embarazo o post parto, retraso mental inexplicable o historia de glutamina o amonio plasmático elevado; 2) aquellos con letargo y vómitos cíclicos, episodios de irritabilidad extrema, ataxia, BUN bajo, que evitan las proteínas; 3) aquellos con una historia familiar de TCU o una historia familiar de muertes de niños inexplicables (particularmente hombres); 4) aquellos con otros signos o síntomas de TCU. Pacientes que desarrollan síntomas de encefalopatía hiperamonémica inexplicable mientras reciben terapia con valproato deben recibir tratamiento rápido (incluyendo discontinuación de la terapia con valproato) y ser evaluado por trastornos del ciclo de la urea subyacente (ver Contraindicaciones y Precauciones). Conducta e Ideación Suicida: Se ha reportado un aumento en el riesgo de pensamientos o conducta suicida en pacientes que toman drogas antiepilépticas (AEDs) para cualquier indicación. El aumento del riesgo de pensamientos o conducta suicida con AEDs se observó tan temprano como una semana después del inicio del tratamiento con AEDs y persistió por la duración del tratamiento evaluado. El riesgo relativo para pensamientos o conducta suicida fue mayor en estudios clínicos para epilepsia que en estudios clínicos para psiquiátricos u otras condiciones, pero las diferencias de riesgo absoluto fueron similares para las indicaciones de epilepsia y psiquiátricas. Los pacientes tratados con un AED por cualquier indicación se deben monitorear para la emergencia o empeoramiento de depresión, pensamientos o conducta suicida, y/o cualquier cambio inusual en el ánimo o conducta. Cualquiera que considere prescribir divalproato de sodio o cualquier otro AED debe balancear el riesgo de pensamientos o conducta suicida con el riesgo de no tratar la enfermedad. La epilepsia y muchas otras enfermedades para las cuales se prescriben AED, están ellas mismas asociadas con morbilidad y un aumento del riesgo de pensamientos o conducta suicida. Si durante el tratamiento emergen pensamientos y conductas suicidas, el prescriptor necesita considerar si la emergencia de estos síntomas en cualquier paciente se puede relacionar a la enfermedad que está siendo tratada. Se debe informar a los pacientes, sus cuidadores y familiares que los AEDs aumentan el riesgo pensamientos y conducta suicida, y deben ser aconsejados sobre la necesidad de estar alertas a la emergencia o empeoramiento de signos y síntomas de depresión, cualquier cambio inusual en el ánimo o conducta, o la emergencia de pensamientos, conducta suicidas, o pensamiento acerca de autoagresión. Conductas de preocupación se deben reportar inmediatamente al personal de salud. Interacción con Antibióticos Carbapenem: Los antibióticos carbapenem (ertapenem, imipenem, meropenem) pueden reducir las concentraciones séricas de ácido valproico a niveles sub-terapéuticos, produciendo pérdida del control de las convulsiones. Las concentraciones séricas de ácido valproico se deben monitorear frecuentemente después del inicio de la terapia con carbapenem. Se deben considerar terapias anticonvulsivantes o antibacterianas alternativas si las concentraciones de ácido valproico caen significativamente o se deteriora el control de las convulsiones (ver Interacciones). Somnolencia en Ancianos: En un estudio multicéntrico, doble ciego de valproato en pacientes ancianos con demencia (edad media = 83 años), las dosis se incrementaron en 125 mg/día a una dosis objetivo de 20 mg/kg/día. Una proporción significativamente mayor de pacientes con valproato presentó somnolencia en comparación con placebo, y aunque no fue estadísticamente significativa, se observó una mayor proporción de pacientes con deshidratación. Las discontinuaciones por somnolencia también fueron significativamente superiores a las observadas con placebo. En algunos pacientes con somnolencia (aproximadamente la mitad), se asoció con una menor ingesta alimenticia y pérdida de peso. Existió una tendencia entre los pacientes que experimentaron estos eventos a presentar una menor concentración basal de albúmina, menor clearance de valproato y un BUN más alto. En los pacientes mayores, la dosificación se debe incrementar más lentamente y con monitoreo regular de la ingesta de líquidos y alimentos, deshidratación, somnolencia y otros eventos adversos. Se debe considerar reducciones de la dosis o discontinuación de valproato en pacientes con una menor ingesta de alimentos o líquidos y en pacientes con excesiva somnolencia (ver Dosificación). Trombocitopenia: La frecuencia de efectos adversos (especialmente niveles elevados de enzimas hepáticas y trombocitopenia (ver Precauciones), puede ser dosis-relacionada. En un estudio clínico de Divalproato de Sodio como monoterapia en pacientes con epilepsia, 34/126 pacientes (27%) que recibieron aproximadamente 50 mg/kg/día en promedio, presentaron al menos un valor de plaquetas ≤ 75 x 109/L. Aproximadamente la mitad de estos pacientes discontinuaron el tratamiento, retornando el recuento de plaquetas a niveles normales. En los pacientes restantes, el recuento de plaquetas se normalizó mientras se continuaba el tratamiento. En este estudio, la probabilidad de trombocitopenia pareció aumentar significativamente en concentraciones totales de valproato de ≥ 110 mcg/mL (mujeres) o ≥ 135 mcg/mL (hombres). El beneficio terapéutico que puede acompañar a las dosis más altas debe por tanto ser sopesado contra la posibilidad de una mayor incidencia de efectos adversos. Uso en Embarazo: De acuerdo a los informes publicados y no publicados, el ácido valproico puede producir efectos teratogénicos, tales como defectos del tubo neural (por ejemplo, espina bífida) en los recién nacidos de mujeres que reciben la droga durante el embarazo. Hay datos que sugieren un aumento en la incidencia de malformaciones congénitas asociadas al uso del ácido vlproico durante el embarazo en comparación con algunas otras drogas antiepilépticas. Por lo tanto, el ácido valproico se debe considerar para las mujeres en edad fértil solo después de haber discutido los riesgos a fondo con la paciente y pesado contra los beneficios potenciales del tratamiento. Los datos descritos abajo fueron obtenidos casi exclusivamente de mujeres quienes recibieron valproato para tratar epilepsia. Existen múltiples reportes en la literatura clínica que indican que el uso de drogas antiepilépticas durante el embarazo resulta en una mayor incidencia de defectos de nacimiento en los recién nacidos. Por lo tanto, las drogas antiepilépticas se deben administrar a mujeres en edad fértil sólo si han demostrado ser esenciales para el manejo de su enfermedad. Los datos descritos abajo fueron obtenidos casi exclusivamente de mujeres que recibieron valproato para tratar la epilepsia La incidencia de defectos del tubo neural en el feto puede verse aumentada en mujeres que reciben valproato durante el primer trimestre del embarazo. El Centro para Control de Enfermedades de los Estados Unidos (CDC) ha estimado que el riesgo de las mujeres expuestas a ácido valproico de tener hijos con espina bífida es de aproximadamente 1 a 2%. Se han reportado otras anomalías congénitas (por ejemplo, defectos craneofaciales, malformaciones cardiovasculares y anomalías que comprometen diferentes sistemas corporales), compatibles e incompatibles con la vida. No se dispone de datos suficientes para determinar la incidencia de estas anomalías congénitas. La mayor incidencia de anomalías congénitas en mujeres tratadas con drogas antiepilépticas con trastornos convulsivos no puede considerarse como una relación de causa y efecto. Existen problemas metodológicos intrínsecos para la obtención de datos adecuados sobre la teratogenicidad de drogas en seres humanos; los factores genéticos o la condición epiléptica en si misma, pueden ser más importantes que la terapia farmacológica en la contribución a las anomalías congénitas. Ha habido informes de retraso en el desarrollo, autismo y/o trastornos del espectro del autismo en los recién nacidos de mujeres que han estado expuestas al ácido valproico durante el embarazo. Las pacientes que reciben valproato pueden desarrollar alteraciones de la coagulación. Una paciente que presentaba bajos niveles de fibrinógeno mientras recibía múltiples anticonvulsivantes incluyendo valproato dio luz a un lactante con afibrinogenemia quien posteriormente falleció de hemorragia. Si se utiliza valproato durante el embarazo, se deben monitorear cuidadosamente los parámetros de coagulación. Se ha reportado insuficiencia hepática, resultando en la muerte de un recién nacido y de un lactante, a continuación del uso de valproato durante el embarazo. Se deben considerar exámenes para detector defectos del tubo neural y otros usando procedimientos actualmente aceptados, como parte de la rutina pre-natal en mujeres embarazadas que reciben valproato. Las drogas antiepilépticas no se deben discontinuar abruptamente en pacientes en quienes la droga se administra para prevenir convulsiones mayores debido a la fuerte posibilidad de precipitar un status epiléptico con hipoxia asociada y riesgo de vida. En casos individuales donde la severidad y frecuencia del trastorno convulsivo son tales que la remoción de la dosis del medicamento no lo expone a un riesgo serio para el paciente, la discontinuación de la droga se puede considerar previo a y durante el embarazo, aunque no se puede decir con confianza que aún convulsiones menores no lo expone a algún riesgo para el desarrollo del embrión o feto. Los estudios en animales han demostrado teratogenicidad inducida por valproato. Se ha observado un incremento en la frecuencia de malformaciones, así como también retardo del crecimiento intrauterino y muerte en ratones, ratas, conejos y monos a continuación de la exposición prenatal al valproato. Las malformaciones del sistema esquelético son las anormalidades estructurales más comúnmente observadas en los animales de experimentación, aunque se han observado defectos de cierre del tubo neural en ratones expuestos a concentraciones plasmáticas maternas de valproato que excedieron de 230 mcg/mL (2.3 veces el límite superior del rango terapéutico para la epilepsia en humanos) durante períodos susceptibles del desarrollo embrionario. La administración de una dosis oral de 200 mg/kg/día o más (50% de la dosis diaria humana máxima o más en una base de mg/m2) a ratas preñadas durante organogénesis produjo malformaciones (esqueléticas, cardíacas y urogenitales) y retardo del crecimiento en las crías. Estas dosis resultaron en niveles plasmáticos máximos maternos de valproato de aproximadamente 340 mcg/mL o más (3.4 veces el límite superior del rango terapéutico para la epilepsia en humanos o más). Se han reportado déficit conductuales en las crías de ratas que recibieron una dosis de 200 mg/kg/día a través de gran parte del embarazo. Una dosis oral de 350 mg/kg/día (aproximadamente el doble de la dosis diaria humana máxima en una base de mg/m2) produjo malformaciones esqueléticas y viscerales en conejos expuestos durante organogénesis. Las malformaciones esqueléticas, retardo del crecimiento y muerte se observaron en monos rhesus a continuación de la administración de una dosis oral de 200 mg/kg/día (igual a la dosis diaria humana máxima en una base de mg/m2) durante organogénesis. Esta dosis resultó en niveles plasmáticos maternos máximos de valproato de aproximadamente 280 mcg/mL (2.8 veces el límite superior del rango terapéutico para la epilepsia en seres humanos).
Interacciones.
Efectos de las Drogas Co-administradas sobre el Clearance de Valproato: Las drogas que afectan el nivel de expresión de las enzimas hepáticas, especialmente aquellas que elevan los niveles de las glucuronosiltransferasas, pueden incrementar el clearance de valproato. Por ejemplo, la fenitoína, carbamazepina, y fenobarbital (o primidona) puede duplicar el clearance de valproato. Por ello, los pacientes bajo monoterapia generalmente presentarán vidas medias más prolongadas y concentraciones más altas que los pacientes que están recibiendo politerapia con drogas antiepilépticas. Por el contrario, se puede esperar que las drogas que son inhibidoras de las isoenzimas del citocromo P450, por ejemplo, los antidepresivos, ejerzan poco efecto sobre el clearance de valproato dado que la oxidación del citocromo P450 mediada microsomalmente es una vía metabólica secundaria relativamente menor en comparación con la glucuronidación y la beta-oxidación. Debido a estos cambios en el clearance de valproato, se debe incrementar el monitoreo de las concentraciones de valproato y de las drogas concomitantes cada vez que se introducen o discontinúan drogas inductoras de las enzimas. El siguiente listado entrega información acerca de la influencia potencial sobre la farmacocinética de valproato de numerosos fármacos de prescripción frecuente. La lista no es exhaustiva, y no puede serlo, dado que continuamente se reportan nuevas interacciones. Drogas para las Cuales se ha Observado Interacción Potencialmente Importante: Aspirina - Un estudio que involucró la administración conjunta de aspirina a dosis antipiréticas (11 a 16 mg/kg) con valproato a pacientes pediátricos (n= 6) reveló una reducción en la unión a proteínas y una inhibición del metabolismo de valproato. La fracción libre del valproato se incrementó en cuatro veces en presencia de aspirina en comparación a lo observado para valproato solo. La vía de b-oxidación consistente de 2-E-ácido valproico, 3-OH-ácido valproico, y 3-ceto ácido valproico se redujo desde un 25% de metabolitos totales excretados con valproato solo a 8.3% en presencia