VALIXA®
ROCHE
Antivírico.
Composición.
Principio activo: valganciclovir (en forma de clorhidrato de valganciclovir). Comprimidos recubiertos de 450 mg. Excipientes: Comprimidos recubiertos: Núcleo de los comprimidos: povidona, crospovidona, celulosa microcristalina, ácido esteárico en polvo. Recubrimiento de los comprimidos: hidroxipropilmetilcelulosa, dióxido de titanio (E171), polietilenglicol, óxido de hierro rojo (E172), polisorbato. Forma farmacéutica: Comprimidos recubiertos. Vía de administración: Oral. Declaración de esterilidad / radiactividad: No procede.
Farmacología.
Propiedades farmacodinámicas: Mecanismo de acción: El valganciclovir es un L-valil-éster (profármaco) del ganciclovir, que, administrado por vía oral, se transforma rápidamente en ganciclovir por la acción de estearasas intestinales y hepáticas. El ganciclovir es un análogo sintético de la 2'-desoxiguanosina, que actúa inhibiendo in vitro e in vivo la replicación de los virus herpéticos. Los virus humanos sensibles son los siguientes: citomegalovirus (CMV) humano, virus del herpes simple tipos 1 y 2, virus del herpes humano tipos 6, 7 y 8, virus de Epstein-Barr, virus de la varicela-zóster y virus de la hepatitis B. En las células infectadas por CMV, el ganciclovir es fosforilado inicialmente a monofosfato de ganciclovir por la proteincinasa vírica UL97. A continuación, las cinasas celulares lo fosforilan a trifosfato de ganciclovir, que se metaboliza lentamente después en el interior celular. Este efecto se ha demostrado en células infectadas por virus del herpes simple y CMV humano, con una semivida de, respectivamente, 18 horas y entre 6 y 24 horas tras la eliminación del ganciclovir extracelular. Dado que la fosforilación depende en gran medida de la cinasa vírica, la del ganciclovir tiene lugar preferentemente en las células infectados por los virus. La actividad virustática del ganciclovir obedece a la inhibición de la síntesis del ADN vírico por: a) la inhibición competitiva de la incorporación del trifosfato de desoxiguanosina al ADN por la polimerasa del ADN vírico, y b) la incorporación del trifosfato de ganciclovir al ADN vírico, deteniéndose así total o casi totalmente la elongación del ADN vírico. La CI50 antivírica típica frente al CMV in vitro suele oscilar entre 0,08 mM (0,02 mg/ml) y 14 mM (3,5 mg/ml). Clínicamente, el efecto antivírico de Valixa ha quedado demostrado en el tratamiento de pacientes con sida que sufrían retinitis citomegalovírica recién diagnosticada (estudio clínico WV15376). La eliminación de CMV disminuyó desde el 46% (32 de 69 pacientes) al comienzo del estudio al 7% (4/55) después de cuatro semanas de tratamiento con Valixa. Estudios clínicos / Eficacia: Tratamiento de la retinitis citomegalovírica: Se han realizado estudios clínicos de Valixa en pacientes con sida y retinitis citomegalovírica. En el tratamiento de inducción de la retinitis citomegalovírica, Valixa ha demostrado una eficacia comparable a la del ganciclovir i.v. A los pacientes con retinitis citomegalovírica recién diagnosticada de un estudio se los asignó aleatorizadamente al tratamiento de inducción con Valixa o ganciclovir i.v. Al cabo de 4 semanas, la proporción de pacientes con progresión de la retinitis citomegalovírica era idéntica en ambos grupos. Tras el tratamiento de inducción, los pacientes de este estudio recibieron tratamiento de mantenimiento con Valixa, en una dosis de 900 mg/día. La duración media (mediana) del tiempo transcurrido entre la randomización y la progresión de la retinitis citomegalovírica fue de 226 (160) días en el grupo que recibió Valixa como tratamiento de inducción y de mantenimiento y de 219 (125) días en el que recibió ganciclovir i.v. como tratamiento de inducción y Valixa como tratamiento de mantenimiento. Con Valixa, la exposición sistémica al ganciclovir es similar a la alcanzada con las dosis recomendadas de ganciclovir i.v., cuya eficacia en el tratamiento de la retinitis citomegalovírica está demostrada. Entre el ABC de ganciclovir y el tiempo transcurrido hasta la progresión de la retinitis citomegalovírica existe correlación. Prevención de la infección citomegalovírica en los trasplantes: Se ha realizado un estudio de doble-ciego y doble enmascaramiento, con tratamiento comparativo activo, en pacientes sometidos a trasplante de corazón, hígado o riñón con alto riesgo de infección citomegalovírica (D+/R-), que recibieron Valixa (900 mg una vez al día) o ganciclovir p.o. (1.000 mg tres veces al día), empezando la administración dentro de los 10 días siguientes al trasplante y manteniéndola hasta el día 100 después del trasplante. Según la valoración del Comité de Evaluación, la incidencia de infección citomegalovírica (síndrome citomegalovírico + citomegalovirosis histoinvasora) durante los 6 primeros meses tras el trasplante fue del 12,1% en el grupo de Valixa (n = 239), frente a un 15,2% en el grupo de ganciclovir p.o. (n = 125). La gran mayoría de los casos se produjeron tras la terminación de la profilaxis (día 100° postrasplante), y, en general, los del grupo de valganciclovir después que los del grupo de ganciclovir p.o. La incidencia de rechazos agudos durante los 6 primeros meses fue del 29,7% entre los pacientes que recibieron valganciclovir, frente al 36,0% en los del grupo de ganciclovir p.o. En los receptores de un trasplante renal de alto riesgo, la extensión de la profilaxis anticitomegalovírica con Valixa hasta el día 200 postrasplante demostró tener un mejor efecto preventivo de la infección citomegalovírica dentro de los 12 primeros meses postrasplante que el régimen de 100 días. Se ha realizado un estudio de doble-ciego, controlado con placebo, en 326 receptores de un trasplante renal con alto riesgo de infección citomegalovírica (D+/R-) para evaluar la eficacia y la seguridad de extender la profilaxis anticitomegalovírica con valganciclovir de 100 a 200 días tras el trasplante. Se aleatorizó a los pacientes (1:1) para recibir Valixa en comprimidos (900 mg una vez al día) dentro de los 10 días siguientes al trasplante hasta el día 200 o el día 100 postrasplante, seguido de 100 días de placebo. En la Tabla 6 se muestra la proporción de pacientes que desarrollaron infección citomegalovírica durante los 12 primeros meses tras el trasplante.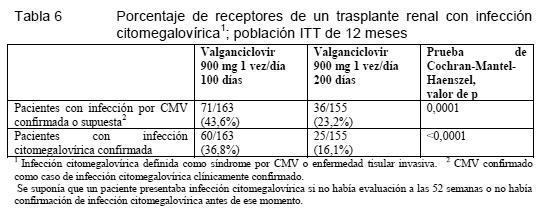
La tasa de supervivencia del injerto a los 12 meses postransplante fue del 98,1% (160/163) con el régimen de 100 días y del 98,2% (152/155) con el de 200 días. La incidencia de rechazo agudo demostrado por biopsia a los 12 meses postransplante fue del 17,2% (28/163) con el régimen de 100 días y del 11,0% (17/155) con el de 200 días. Resistencia vírica: Tras la administración prolongada de valganciclovir puede desarrollarse resistencia vírica al ganciclovir por selección de mutaciones, bien en el gen de la cinasa vírica (UL97) responsable de la monofosforilación del ganciclovir, bien en el gen de la polimerasa vírica (UL54). Un virus con mutaciones en el gen de la UL97 es resistente al ganciclovir solamente, mientras que un virus con mutaciones en el gen de la UL54 puede presentar resistencia cruzada a otros antivíricos de acción sobre la polimerasa vírica, y viceversa. Tratamiento de la retinitis citomegalovírica: El análisis genotípico de cepas de CMV en leucocitos polimorfonucleares aisladas en 148 pacientes con retinitis citomegalovírica de un estudio clínico ha puesto de manifiesto mutaciones de la UL97 en un 2,2%, 6,5%, 12,8% y 15,3% después de 3, 6, 12 y 18 meses, respectivamente, de tratamiento con valganciclovir. Prevención de la infección citomegalovírica en los trasplantes: Se ha estudiado la resistencia mediante el análisis genotípico de CMV en muestras de leucocitos polimorfonucleares recogidos a) el día 100° (final de la profilaxis farmacológica del estudio) y b) hasta 6 meses tras el trasplante en los casos de sospecha de infección citomegalovírica. De los 245 pacientes del grupo de valganciclovir, el día 100° había 198 muestras para el análisis, el cual no reveló ninguna mutación de resistencia al ganciclovir. En los pacientes del grupo comparativo de ganciclovir p.o. se detectaron 2 mutaciones de resistencia al ganciclovir en las 103 muestras analizadas (1,9%). De los 245 pacientes del grupo de valganciclovir, se analizaron muestras de 50 pacientes con sospecha de infección citomegalovírica, pero no se detectó ninguna mutación de resistencia. De los 125 pacientes del grupo comparativo de ganciclovir, se analizaron muestras de 29 pacientes con sospecha de infección citomegalovírica y se detectaron dos mutaciones de resistencia, lo que equivale a una incidencia de resistencia del 6,9%. Propiedades farmacocinéticas: Las propiedades farmacocinéticas del valganciclovir se han estudiado en pacientes seropositivos frente al virus de la inmunodeficiencia humana (VIH) y CMV, pacientes con sida y retinitis citomegalovírica y pacientes sometidos a trasplante de órgano sólido. Los parámetros determinantes de la exposición del ganciclovir formado a partir del valganciclovir son la biodisponibilidad y la función renal. La biodisponibilidad del ganciclovir formado del valganciclovir es comparable en todos los grupos de pacientes estudiados. La exposición sistémica al ganciclovir en los receptores de un trasplante de corazón, riñón o hígado era similar tras la administración p.o. del valganciclovir, de acuerdo con el algoritmo de dosis para pacientes con disfunción renal. Absorción: El valganciclovir es un profármaco del ganciclovir; se absorbe bien en el tubo digestivo y se metaboliza rápidamente a ganciclovir en la pared intestinal y el hígado. La biodisponibilidad absoluta del ganciclovir formado a partir del valganciclovir es del 60% aproximadamente. La exposición sistémica al valganciclovir es transitoria y baja; los valores de ABC24 y Cmáx se hallan en torno al 1% y el 3%, respectivamente, de los registrados con el ganciclovir. La proporcionalidad entre la dosis y el ABC del ganciclovir tras la administración de valganciclovir en dosis de entre 450 y 2.625 mg se ha demostrado únicamente en condiciones de administración con alimentos. Cuando el valganciclovir se administró junto con alimentos en la dosis recomendada de 900 mg, aumentaron los valores medios tanto de ABC24 (30%, aproximadamente) como de Cmáx (14%, aproximadamente) del ganciclovir. Por consiguiente, se recomienda administrar Valixa junto con alimentos (ver Dosificación). Distribución: Dada la rápida conversión del valganciclovir en ganciclovir, no se ha determinado la unión de Valixa a las proteínas. La unión del ganciclovir a las proteínas era del 1-2% en concentraciones de entre 0,5 y 51 mg/ml. Tras la administración i.v. del ganciclovir, el volumen de distribución en equilibrio era de 0,680 ± 0,161 l/kg. Metabolismo: El valganciclovir se hidroliza rápidamente a ganciclovir. No se han detectado otros metabolitos. Ningún metabolito del ganciclovir radiomarcado administrado p.o. (1.000 mg en una dosis única) representaba más del 1-2% de la radiactividad recuperada en las heces o la orina. Eliminación: La excreción renal en la forma de ganciclovir por filtración glomerular y secreción tubular activa constituye la principal vía de eliminación del Valixa. El aclaramiento renal representa el 81,5% ±22% del aclaramiento sistémico del ganciclovir. Farmacocinética en poblaciones especiales: Pacientes con insuficiencia renal: Una función renal decreciente comportaba un descenso del aclaramiento de ganciclovir formado a partir de su precursor el valganciclovir, con el aumento correspondiente de la semivida terminal. Por tanto, es necesario ajustar la dosis en pacientes con la función renal menoscabada (ver Pautas posológicas especiales y Advertencias). Pacientes con insuficiencia hepática: En un estudio abierto y cruzado con 4 grupos (n = 28), se estudió la farmacocinética del valganciclovir en receptores de trasplante hepático en estado estable. Tras una dosis única de 900 mg de valganciclovir tomada con alimentos, la biodisponibilidad absoluta del ganciclovir formado a partir del valganciclovir era del 60% aproximadamente, lo que concuerda con las estimaciones en otras poblaciones. El ABC0-24 del ganciclovir era comparable al hallado tras la administración de una dosis de 5 mg/kg de ganciclovir i.v. a pacientes receptores de un trasplante de hígado. Datos preclínicos sobre seguridad: Carcinogenicidad: Estos resultados concuerdan con los obtenidos en un estudio de ganciclovir que revelaban el poder cancerígeno de este fármaco en el ratón. Como el ganciclovir, el valganciclovir puede ser cancerígeno. Mutagenicidad: El valganciclovir y el ganciclovir eran mutágenos en células de linfoma murino y clastógenos en células de mamífero. Trastornos de la fecundidad: En los animales, el ganciclovir provoca infertilidad y es teratógeno. Con el valganciclovir no se han realizado estudios de toxicidad en la reproducción, dada su rápida y amplia transformación en ganciclovir. A este respecto, la misma advertencia es válida para ambos fármacos (ver Advertencias). Teniendo en cuenta la aspermatogenia observada en estudios con animales tras la exposición sistémica al ganciclovir en concentraciones inferiores a las terapéuticas, se estima que es probable que el ganciclovir (y el valganciclovir) inhiban la formación de espermatozoides en el hombre. Teratogenicidad: En los animales, el ganciclovir es teratógeno. Con el valganciclovir no se han realizado estudios de toxicidad en la reproducción, dada su rápida y amplia transformación en ganciclovir. A este respecto, la misma advertencia es válida para ambos fármacos (ver Advertencias). Los datos obtenidos ex vivo con un modelo de placenta humana muestran que el ganciclovir atraviesa la barrera placentaria y que el mecanismo más probable de entrada es el de simple difusión. El proceso era no saturable en una concentración de 1 a 10 mg/ml y se desarrollaba por difusión pasiva.
Indicaciones.
Valixa está indicado para el tratamiento de la retinitis citomegalovírica (retinitis por citomegalovirus, CMV) en pacientes con el síndrome de inmunodeficiencia adquirida (sida). Valixa está indicado para la prevención de la infección citomegalovírica (infección/enfermedad por CMV) en pacientes de alto riesgo sometidos a trasplante de riñón, corazón o páncreas-riñón.
Dosificación.
¡Atención!: Para evitar una sobredosis, es fundamental seguir estrictamente las recomendaciones posológicas. Dosis habitual: Valixa debe tomarse por vía oral (p.o.), junto con alimentos (ver Farmacocinética en poblaciones especiales y Absorción). Valixa se convierte rápida y ampliamente en ganciclovir, el principio activo. La biodisponibilidad del ganciclovir formado a partir de Valixa es hasta 10 veces mayor que la del ganciclovir oral. Deben respetarse exactamente la posología y la administración de Valixa en comprimidos tal como se señala a continuación (ver Advertencias y Sobredosificación). Adultos: Tratamiento de inducción de la retinitis citomegalovírica: Para los pacientes adultos con retinitis citomegalovírica activa se recomienda una dosis de 900 mg dos veces al día, durante 21 días. Un tratamiento de inducción prolongado puede elevar el riesgo de toxicidad medular (ver Advertencias). Tratamiento de mantenimiento de la retinitis citomegalovírica: Tras el tratamiento de inducción, así como en los pacientes adultos con retinitis citomegalovírica inactiva, se recomienda una dosis de 900 mg una vez al día. En caso de empeoramiento de la retinitis, puede repetirse el tratamiento de inducción (ver Tratamiento de inducción de la retinitis citomegalovírica). Prevención de la infección citomegalovírica en los trasplantes: Para los receptores de un trasplante renal de alto riesgo, la dosis recomendada es de 900 mg una vez al día, empezando dentro de los 10 días siguientes al trasplante y manteniéndola hasta 200 días después del trasplante. Para los pacientes que han recibido un trasplante de órgano sólido que no sea un riñón, la dosis recomendada es de 900 mg una vez al día, empezando dentro de los 10 días siguientes al trasplante y manteniéndola hasta 100 días después del trasplante. Pautas posológicas especiales: Pacientes con insuficiencia renal: Las cifras de creatinina sérica o de aclaramiento de la creatinina han de vigilarse estrechamente. La dosis debe ajustarse para los pacientes adultos en función del aclaramiento de la creatinina como se muestra en las tablas 1 y 2 (ver Farmacocinética en poblaciones especiales y Advertencias).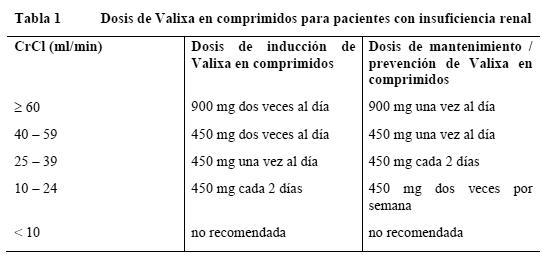
El aclaramiento de la creatinina se calcula a partir de la creatinina sérica por las fórmulas siguientes: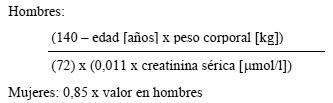
Pacientes con leucocitopenia, neutrocitopenia, anemia, trombocitopenia o pancitopenia graves: Se han descrito leucocitopenia, neutrocitopenia, anemia, trombocitopenia, pancitopenia, depresión medular y anemia aplásica entre los pacientes tratados con Valixa (y ganciclovir). No debe iniciarse el tratamiento si la cifra absoluta de neutrófilos es inferior a 500/ml, el recuento plaquetario se halla por debajo de 25.000/ml o si la concentración de hemoglobina es menor de 8 g/dl (ver Farmacocinética en poblaciones especiales y Reacciones adversas). Ancianos: No se han determinado la inocuidad y la eficacia en esta población. Pacientes pediátricos: No se han realizado estudios clínicos adecuados y bien controlados sobre la seguridad y la eficacia de Valixa en pacientes pediátricos.
Contraindicaciones.
Valixa está contraindicado en pacientes alérgicos al valganciclovir, el ganciclovir o cualquier otro componente del producto. Dada la semejanza de la estructura química de Valixa, aciclovir y valaciclovir, puede producirse una reacción cruzada de hipersensibilidad entre estos fármacos.
Reacciones adversas.
Ensayos clínicos: El valganciclovir es un profármaco del ganciclovir. Administrado p.o., se convierte rápidamente en ganciclovir. Por consiguiente, cabe suponer que los conocidos efectos adversos asociados al ganciclovir se producirán también con Valixa. Todos los efectos secundarios descritos en los estudios clínicos con Valixa se habían observado anteriormente con el ganciclovir. Tratamiento de la retinitis citomegalovírica en pacientes con sida: Las características toxicológicas del valganciclovir y el ganciclovir i.v. durante 28 días de estudio aleatorizado (21 días con la dosis de inducción y 7 días con la de mantenimiento) en 79 pacientes eran similares. Las reacciones adversas más frecuentes consistieron en diarrea, neutrocitopenia y fiebre. Hubo más pacientes que refirieron diarrea, candidosis oral, cefalea y fatiga en el grupo con valganciclovir p.o., y viceversa: fue mayor el número de pacientes con náuseas, además de reacciones en el sitio de inyección, en el grupo tratado con ganciclovir i.v. (ver Tabla 3).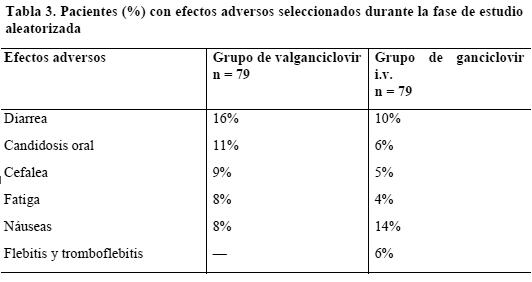
En la Tabla 4 se muestran los acontecimientos adversos -independientemente de la gravedad y la relación con el fármaco- con una incidencia ≥ 5% en los estudios en los que se evaluaba el valganciclovir en la retinitis citomegalovírica o el trasplante de órgano sólido. Los datos de la Tabla 4 correspondientes a la retinitis citomegalovírica se basan en dos estudios clínicos (n = 370) en los que pacientes con esta enfermedad recibieron Valixa en una dosis de 900 mg dos veces al día o una vez al día, lo que corresponde a la pauta de inducción y de mantenimiento, respectivamente. Al 65%, aproximadamente, de estos pacientes se los trató con valganciclovir durante más de nueve meses (la duración máxima fue de 30 meses). Los efectos secundarios notificados con mayor frecuencia (% de pacientes) en estos dos estudios clínicos (n = 370) entre los pacientes tratados con Valixa, independientemente de la gravedad y la relación con el fármaco, fueron diarrea (38%), fiebre (26%), náuseas (25%), neutrocitopenia (24%) y anemia (22%). La mayor parte de los acontecimientos adversos fueron leves o moderados. Los efectos adversos notificados con más frecuencia (% de pacientes), independientemente de la gravedad, que el investigador consideró relacionados (remota, posible o probablemente) con Valixa fueron neutrocitopenia (21%), anemia (14%), diarrea (13%) y náuseas (9%). Prevención de la infección citomegalovírica en los trasplantes: La Tabla 4 recoge las reacciones adversas -independientemente de la gravedad y la relación con el fármaco- con una incidencia ≥ 5% en un ensayo clínico (hasta 28 días después del tratamiento estudiado) en el que pacientes sometidos a trasplante de órgano sólido recibieron valganciclovir (n = 244) o ganciclovir p.o. (n = 126), empezando la administración dentro de los 10 días siguientes al trasplante y manteniéndola hasta el día 100 después del trasplante. Los efectos secundarios notificados con mayor frecuencia (% de pacientes) entre los pacientes que recibieron Valixa en este estudio clínico (n = 244), independientemente de la gravedad y la relación con el fármaco, fueron diarrea (30%), temblor (28%), rechazo del injerto (24%), náuseas (23%), cefalea (22%), edema de las extremidades inferiores (21%), estreñimiento (20%), dorsalgia (20%), insomnio (20%), hipertensión (18%) y vómitos (16%). Estos acontecimientos adversos también se produjeron con el ganciclovir p.o. y con una incidencia comparable. La mayor parte de los acontecimientos adversos fueron leves o moderados. Los efectos adversos observados en el ensayo clínico de trasplante de órgano sólido (régimen de administración de 100 días), pero no en los estudios clínicos sobre la retinitis citomegalovírica, con una frecuencia ≥ 2% consistieron en hipertensión (18%), aumento de la creatinina (10%), trastornos metabólicos -por ejemplo hiperpotasemia (14%)- y disfunción hepática (9%). Estos acontecimientos tuvieron una frecuencia similar con ganciclovir p.o. y se los puede considerar como reflejo del proceso patológico subyacente. En los pacientes sometidos a trasplante de órgano sólido tratados hasta el día 100 postrasplante, los efectos adversos notificados con más frecuencia (% de pacientes), independientemente de la gravedad, que el investigador consideró relacionados (remota, posible o probablemente) con Valixa fueron leucocitopenia (9%), diarrea (7%), náuseas (6%) y neutrocitopenia (5%).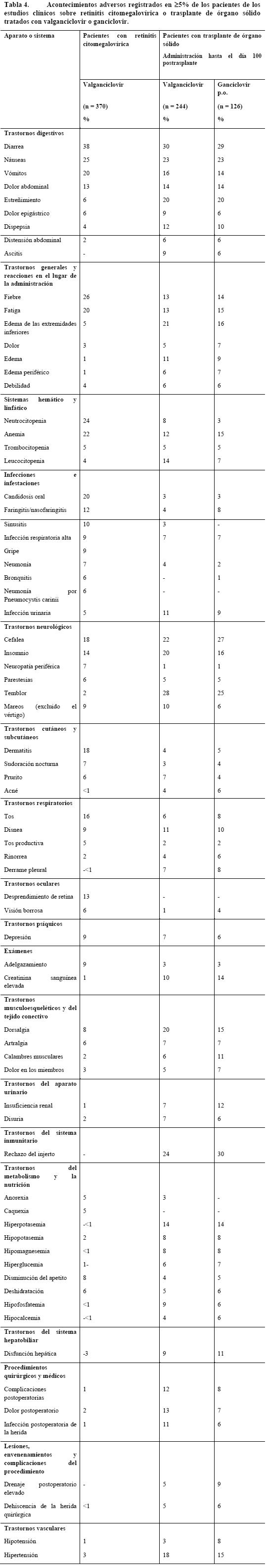
A continuación se consignan los acontecimientos adversos graves notificados en estos tres estudios clínicos (n = 614) que la compañía considera relacionados con Valixa y que tuvieron una frecuencia inferior al 5%, por lo que no se hallan en las dos tablas anteriores: Sistema vascular: pancitopenia, depresión medular, anemia aplásica. Aparato urogenital: disminución del aclaramiento renal de creatinina. Complicaciones hemorrágicas: hemorragia potencialmente mortal asociada a trombocitopenia. Sistema nervioso central y periférico: convulsiones, trastornos psicóticos, alucinaciones, confusión, agitación. Reacciones generales: hipersensibilidad al valganciclovir. La neutrocitopenia grave (RAN < 500/ml) es más frecuente en los pacientes con retinitis citomegalovírica (16%) en tratamiento con valganciclovir que en los sometidos a trasplante de órgano sólido tratados con valganciclovir (5%) o ganciclovir p.o. (3%) hasta el día 100 postransplante. En comparación con los pacientes con retinitis citomegalovírica, el aumento de la creatinina sérica fue mayor en los pacientes con trasplante de órgano tratados hasta el día 100 postransplante, tanto en los que recibieron valganciclovir como en los tratados con ganciclovir p.o. La disfunción renal es una característica común a todos los pacientes sometidos a trasplante de órgano sólido. El perfil global de seguridad del Valixa no varió con la extensión de la profilaxis hasta 200 días en los receptores de un trasplante renal de alto riesgo. Experiencia con ganciclovir: Valixa se convierte rápidamente en ganciclovir. A continuación se indican los efectos secundarios notificados con ganciclovir y no mencionados más arriba: Gastrointestinales: distensión abdominal, colangitis, dispepsia, disfagia, eructos, esofagitis, incontinencia fecal, flatulencia, gastritis, trastorno gastrointestinal, hemorragia gastrointestinal, úlceras bucales, pancreatitis, trastorno lingual. Reacciones generales: ascitis, astenia, infecciones bacterianas, fúngicas y víricas, hemorragia, malestar general, alteración de la mucosa, dolor, reacción de fotosensibilidad, escalofríos, septicemia. Hepáticas: hepatitis, ictericia. Piel y faneras: alopecia, piel seca, sudoración elevada, urticaria. Sistema nervioso central y periférico: sueños anormales, amnesia, ansiedad, ataxia, coma, sequedad de boca, trastornos emocionales, síndrome hipercinético, hipertonía, disminución de la libido, espasmos mioclónicos, nerviosismo, somnolencia, trastornos del pensamiento. Trastornos musculoesqueléticos: artromialgias, síndrome miasténico. Trastornos urogenitales: hematuria, impotencia, insuficiencia renal, polaquiuria. Trastornos del metabolismo y la nutrición: aumento de la fosfatasa alcalina sérica, aumento de la creatincinasa sérica, descenso de glucemia, aumento de la deshidrogenasa láctica, diabetes mellitus, hipoproteinemia. Órganos de los sentidos: ambliopía, ceguera, otalgia, hemorragia ocular, dolor ocular, sordera, glaucoma, disgeusia, acúfenos, visión anormal, alteración del vítreo. Trastornos hemáticos y linfáticos: eosinofilia, leucocitosis, linfadenopatía, esplenomegalia. Trastornos cardiovasculares: arritmias (arritmia ventricular inclusive), migraña, flebitis, taquicardia, tromboflebitis profunda, vasodilatación. Trastornos respiratorios: congestión sinusal. .1 Alteraciones analíticas: En la Tabla 5 se señalan las alteraciones analíticas notificadas con valganciclovir: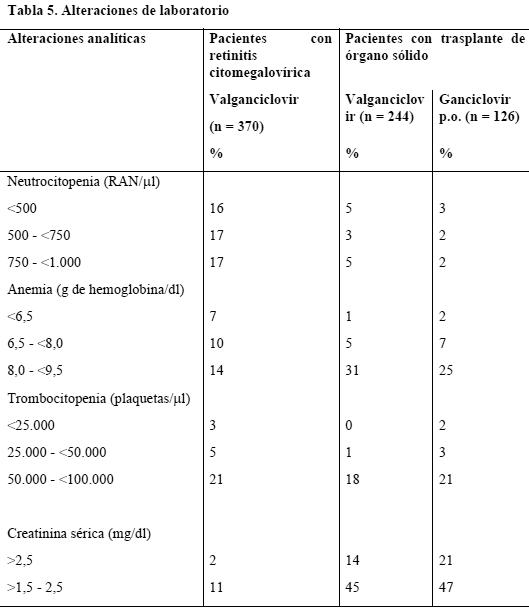
Poscomercialización: Experiencia con ganciclovir: A continuación se indican los efectos secundarios notificados espontáneamente tras la comercialización del ganciclovir i.v. y p.o., no mencionados en ninguno de los apartados anteriores y de los que no cabe excluir una relación causal. Dado que Valixa se convierte rápida y ampliamente en ganciclovir, estos efectos adversos también pueden presentarse tras la administración de Valixa. Anafilaxia. Disminución de la fertilidad masculina. Los acontecimientos adversos notificados tras la comercialización son similares a los previamente observados en los estudios clínicos con Valixa y ganciclovir.
Advertencias.
En la experimentación animal, el ganciclovir fue mutágeno, teratógeno, aspermatógeno y cancerígeno. Por consiguiente, Valixa debe considerarse como potencialmente teratógeno y cancerígeno para el ser humano (ver Instrucciones especiales de uso, manipulación y eliminación). Se estima probable asimismo que Valixa inhiba transitoria o permanentemente la espermatogenia (ver Teratogenicidad, Embarazo y Reacciones adversas). Se han descrito leucocitopenia, neutrocitopenia, anemia, trombocitopenia, pancitopenia, depresión medular y anemia aplásica entre los pacientes tratados con Valixa (y ganciclovir). No debe iniciarse el tratamiento si la cifra absoluta de neutrófilos es inferior a 500/ml, el recuento plaquetario se halla por debajo de 25.000/ml o si la concentración de hemoglobina es menor de 8 g/dl (ver Pautas posológicas especiales, Advertencias y Reacciones adversas). Se recomienda vigilar el hemograma y la cifra de trombocitos durante el tratamiento. En pacientes con leucocitopenia, neutrocitopenia, anemia o trombocitopenia graves, se recomienda considerar la idoneidad del tratamiento con factores de crecimiento hematopoyético y/o la suspensión de la administración (ver Pautas posológicas especiales y Reacciones adversas). Se han descrito convulsiones en pacientes tratados con imipenem+cilastatina y ganciclovir. Valixa no debe utilizarse junto con imipenem+cilastatina, salvo que los beneficios esperados sean mayores que los riesgos (ver Interacciones). Tanto la zidovudina como Valixa pueden provocar neutrocitopenia y anemia. Es posible que algunos pacientes no toleren su administración concomitante en dosis plenas (ver Interacciones). Dado que las concentraciones plasmáticas de didanosina pueden elevarse durante la coadministración con Valixa, es preciso vigilar el desarrollo de la toxicidad de la didanosina durante el tratamiento (ver Interacciones). El uso concomitante con Valixa de otros fármacos mielodepresores o asociados con insuficiencia renal puede incrementar su toxicidad (ver Interacción con otros medicamentos y otras formas de interacción). Drogadicción y dependencia: No hay información disponible sobre drogadicción y dependencia con Valixa. Efectos sobre la capacidad para conducir y utilizar máquinas: Se han descrito convulsiones, sedación, mareos, ataxia y confusión durante el tratamiento con Valixa o ganciclovir. Tales efectos pueden afectar a las actividades que requieren plena atención, como conducir vehículos y manejar máquinas. Pruebas de laboratorio: No hay información disponible sobre pruebas de laboratorio con Valixa.
Interacciones.
Interacciones con Valixa: En un modelo in situ de permeabilidad intestinal en la rata, no se produjo ninguna interacción del valganciclovir con el valaciclovir, la didanosina, el nelfinavir, la ciclosporina, el omeprazol ni el micofenolato mofetilo. Teniendo en cuenta que Valixa se metaboliza a ganciclovir, hay que suponer que las interacciones asociadas al ganciclovir se producirán también con Valixa. Interacciones con ganciclovir: Tan sólo el 1-2% del ganciclovir se une a las proteínas plasmáticas; por ello, no es probable que se produzcan interacciones medicamentosas por desplazamiento del sitio de unión. Imipenem/cilastatina: Se han descrito convulsiones en pacientes tratados simultáneamente con imipenem/cilastatina y ganciclovir. Estos fármacos no deben utilizarse concomitantemente, salvo que los beneficios esperados sean mayores que los riesgos (ver Advertencias). Probenecida: La administración de probenecida con ganciclovir p.o. redujo el aclaramiento renal del ganciclovir en grado estadísticamente significativo (20%), lo que se tradujo en un aumento de la exposición también estadísticamente significativo (40%). Estos cambios podían explicarse por un mecanismo de interacción medicamentosa con competición por los mecanismos de excreción tubular. Por esta razón, es necesario vigilar estrechamente si se producen efectos adversos asociados al ganciclovir en los pacientes tratados con probenecida y Valixa. Zidovudina: Cuando se administró zidovudina con ganciclovir p.o., aumentó el área bajo la curva de concentraciones plasmáticas (ABC) de la zidovudina poco (17%), pero en grado estadísticamente significativo. Se observó también una tendencia hacia concentraciones más bajas de ganciclovir tras su administración con zidovudina, pero sin que la diferencia alcanzara significación estadística. Ahora bien, dado que tanto la zidovudina como el ganciclovir pueden causar neutrocitopenia y anemia, es posible que algunos pacientes no toleren su administración conjunta en dosis plenas (ver Advertencias). Didanosina: La concentración plasmática de didanosina aumentaba sistemáticamente cuando se administraba con ganciclovir, tanto por vía intravenosa (i.v.) como p.o. Con dosis orales de ganciclovir de 3 y 6 g/día, aumentaba el ABC de la didanosina entre un 84% y un 124%. De igual modo, tras dosis i.v. de 5 y 10 mg/kg/día de ganciclovir, el ABC de la didanosina se elevaba entre el 38% y el 67%. Este incremento no es explicable por competición por los mecanismos de secreción tubular, puesto que había un aumento porcentual de la dosis de didanosina excretada. El origen de este incremento podría hallarse en un aumento de la biodisponibilidad o una disminución del metabolismo. Las concentraciones de ganciclovir no experimentaron ningún cambio importante desde un punto de vista clínico. Considerando, sin embargo, el aumento de las concentraciones plasmáticas de didanosina en presencia de ganciclovir, se debe vigilar estrechamente a los pacientes para detectar si se desarrollan efectos adversos de la didanosina (ver Advertencias). Micofenolato mofetilo: Considerando los resultados obtenidos en un estudio de dosis única con micofenolato mofetilo p.o. y ganciclovir i.v. en las dosis recomendadas, así como los efectos conocidos de la insuficiencia renal en la farmacocinética del micofenolato mofetilo y del ganciclovir, cabe prever que la administración simultánea de estos dos fármacos -que pueden competir por los mecanismos de la secreción tubular- incrementará las concentraciones de glucurónido fenólico del ácido micofenólico y ganciclovir. No es de esperar ningún cambio importante en la farmacocinética del ácido micofenólico, y tampoco es preciso ajustar la dosis de micofenolato mofetilo. Los pacientes con insuficiencia renal a los que se administren conjuntamente micofenolato mofetilo y ganciclovir requieren la aplicación de las recomendaciones posológicas para el ganciclovir, así como una estrecha vigilancia. Zalcitabina: La zalcitabina elevaba el ABC0-8 del ganciclovir p.o. en un 13%. Ninguno de los parámetros farmacocinéticos evaluados sufrió ningún cambio con significación estadística. Además, no hubo variaciones farmacocinéticas de la zalcitabina clínicamente importantes en presencia de ganciclovir p.o., aunque sí se observó un pequeño incremento de la velocidad de eliminación. Estavudina: Tras la administración conjunta de estavudina y ganciclovir p.o., no se observó ninguna interacción farmacocinética estadísticamente significativa. Trimetoprima: La trimetoprima reducía en grado estadísticamente significativo el aclaramiento renal del ganciclovir p.o. en un 16,3%, lo que se acompañaba de un descenso también significativo de la velocidad de eliminación terminal, con el correspondiente aumento de la semivida en un 15%. No es probable, sin embargo, que estos cambios tengan importancia clínica, dado que el ABC0-8 y la Cmáx permanecieron inmodificados. La única variación farmacocinética con significación estadística de la trimetoprima coadministrada con ganciclovir consistió en un aumento del 12% de la Cmín. Ahora bien, dado que este cambio no tiene probablemente importancia clínica, no se recomienda ningún ajuste posológico. Ciclosporina: La comparación de las concentraciones mínimas de ciclosporina no mostraba que el ganciclovir afectara a la farmacocinética de la ciclosporina. Sin embargo, hubo algún indicio de aumento de la concentración sérica máxima de creatinina tras el inicio del tratamiento con ganciclovir. Otras interacciones medicamentosas posibles: La toxicidad puede aumentar cuando se administra el ganciclovir con otros fármacos mielodepresores o asociados a trastornos renales (por ejemplo dapsona, pentamidina, flucitosina, vincristina, vinblastina, adriamicina, anfotericina B, análogos nucleosídicos e hidroxiurea). Por consiguiente, estos fármacos sólo deben utilizarse junto con valganciclovir cuando los beneficios esperados sean mayores que los riesgos (ver Advertencias). Uso en poblaciones especiales: Embarazo: Con el valganciclovir no se han realizado estudios de toxicidad en la reproducción, dada su rápida y amplia transformación en ganciclovir. En los animales, el ganciclovir provoca infertilidad y es teratógeno (ver Trastornos de la fertilidad y Teratogenicidad). A las mujeres en edad de procrear se les debe aconsejar que utilicen un método anticonceptivo eficaz durante el tratamiento. De igual modo, a los pacientes de sexo masculino se les aconsejará que utilicen un método anticonceptivo de barrera mientras dure el tratamiento con Valixa y durante un mínimo de 90 días después de concluido (ver Advertencias y Teratogenicidad). No se ha determinado aún la inocuidad de Valixa en mujeres embarazadas. Valixa no debe administrarse a las embarazadas, salvo que el beneficio esperado para la madre sea mayor que el riesgo para el feto. Parto: No hay información disponible sobre el uso de Valixa durante el parto. Lactancia: No se ha estudiado el desarrollo perinatal y posnatal con valganciclovir o ganciclovir, pero no cabe descartar la posibilidad de que el ganciclovir pase a la leche materna y provoque reacciones adversas graves en los lactantes. Por consiguiente, debe decidirse entre suspender la lactancia materna o suspender el tratamiento, según la importancia de Valixa para la madre. Uso en pediatría: No se han realizado estudios clínicos adecuados y bien controlados sobre la seguridad y la eficacia de Valixa en pacientes pediátricos. Uso en geriatría: No se han estudiado la seguridad y la eficacia de Valixa en pacientes ancianos (ver Pautas posológicas especiales). Insuficiencia renal: En pacientes con insuficiencia renal ha de ajustarse la dosis en función del aclaramiento de la creatinina (ver Pautas posológicas especiales y Farmacocinética en poblaciones especiales). Insuficiencia hepática: No hay información disponible sobre el uso de Valixa en pacientes con insuficiencia hepática.
Conservación.
Comprimidos: Los comprimidos deben conservarse a temperatura ambiente (15-30°C). Instrucciones especiales de uso, manipulación y eliminación: Conservación: Los comprimidos no deben romperse ni triturarse. Dado que Valixa está considerado como potencialmente teratógeno y cancerígeno para el ser humano, el manejo de comprimidos rotos o del polvo para solución oral de Valixa exige precaución (ver Advertencias). Evítese el contacto directo de comprimidos rotos o triturados, así como del polvo para la solución y de la solución reconstituida, con la piel o las mucosas. En caso de contacto, lávese la parte afectada con abundante agua y jabón; los ojos deben enjuagarse a fondo con agua estéril o agua corriente si no hay disponible agua estéril. Este medicamento sólo deberá utilizarse hasta la fecha de caducidad, indicada con EXP en el envase. Eliminación de medicamentos no utilizados/caducados: La emisión de productos farmacéuticos al medio ambiente debe reducirse a un mínimo. Los medicamentos no deben eliminarse a través de las aguas residuales, y su eliminación con los residuos domésticos también debe evitarse. Utilice los sistemas de recogida establecidos si los hay en su localidad.
Sobredosificación.
Experiencia con valganciclovir: Un adulto sufrió aplasia medular fatal después de varios días de administración de dosis por lo menos 10 veces superiores a la recomendada para el grado de disfunción renal del paciente (aclaramiento de la creatinina reducido). Una sobredosis de valganciclovir también podría incrementar la toxicidad renal (ver Advertencias y Dosificación). La hemodiálisis y la hidratación pueden resultar útiles para reducir las concentraciones plasmáticas en caso de sobredosis de valganciclovir (ver Farmacocinética en poblaciones es