VIDEX EC
BMS
Antirretroviral.
Descripción.
VIDEX® EC (didanosina) es la marca comercial para didanosina (ddI), un análogo sintético de un nucleósido de purina activo contra el Virus de la Inmunodeficiencia Humana (VIH-1). VIDEX® EC (didanosina) Cápsulas con Gránulos Recubiertos de Liberación Retardada, que contienen gránulos con cubierta entérica, están disponibles para la administración oral en concentraciones de 125, 200, 250 y 400 mg de didanosina. Los ingredientes inactivos en los gránulos incluyen carboximetilcelulosa sódica 12, dietil ftalato, copolímero del ácido metacrílico, hidróxido de sodio, glicolato de almidón de sodio y talco. Las conchas de la cápsula contienen gelatina y dióxido de titanio. Las cápsulas están impresas con tintas comestibles. Didanosina también está disponible en una formulación en polvo. Para información adicional, por favor, consulte la información de prescripción para VIDEX® Polvo Pediátrico para Solución Oral. El nombre químico para la didanosina es 2',3'-dideoxiinosina. La fórmula estructural es: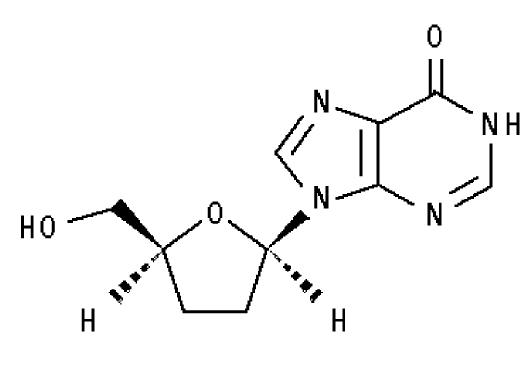
La didanosina es un polvo blanco cristalino con la fórmula molecular C10H12N4O3 y un peso molecular de 236,2. La solubilidad acuosa de la didanosina a 25°C y a un pH de aproximadamente 6, es 27,3 mg/mL. La didanosina es inestable en soluciones acídicas. Por ejemplo, a pH < 3 y a 37°C, 10% de la didanosina se descompone a hipoxantina en menos de 2 minutos. En VIDEX® EC, se usa una cubierta entérica para proteger la didanosina de la degradación por el ácido del estómago.
Propiedades.
Microbiología: Mecanismo de Acción: La didanosina es un análogo sintético del nucleósido que ocurre naturalmente, desoxiadenosina, en el cual el grupo 3'-hidroxil es reemplazado por hidrógeno. Intracelularmente, la didanosina es convertida por las enzimas celulares al metabolito activo, 5'-trifosfato de didesoxiadenosina. El 5'-trifosfato de didesoxiadenosina inhibe la actividad de la transcriptasa reversa del VIH-1 tanto por competencia con el sustrato natural, 5'-trifosfato de didesoxiadenosina, como por su incorporación dentro del ADN viral causando la terminación de la elongación de la cadena del ADN viral. Actividad Antiviral en Cultivo de Célula: La actividad anti-VIH-1 de la didanosina fue evaluada en una variedad de líneas celulares linfoblásticas infectadas con VIH-1 y cultivos de células monocitos/macrófagos. La concentración de droga necesaria para inhibir la replicación viral en un 50% (EC50) estaba en un rango de 2,5 a 10 mM (1mM = 0,24 mg/mL) en líneas de células linfoblásticas y de 0,01 a 0,1 mM en cultivos de células monocitos/macrófagos. Resistencia: Los aislados de VIH-1 con sensibilidad reducida a la didanosina han sido seleccionados en cultivo de célula y también fueron obtenidos de pacientes tratados con didanosina. El análisis genético de los aislados de pacientes tratados con didanosina mostró mutaciones en el gen de la transcriptasa reversa que resultaron en sustituciones de los aminoácidos K65R, L74V y M184V. La mutación L74V fue más frecuentemente observada en los aislados clínicos. El análisis fenotípico de los aislados de VIH-1 de 60 pacientes (algunos con tratamiento previo con zidovudina), quienes recibieron de 6 a 24 meses de monoterapia con didanosina demostró que los aislados de 10 a 60 pacientes exhibieron un promedio de disminución de 10 veces en la susceptibilidad a la didanosina en cultivo de célula en comparación con los aislados basales. Los aislados clínicos que mostraron una disminución en la susceptibilidad a la didanosina incluían una o más de las mutaciones asociadas con resistencia a la didanosina. Resistencia Cruzada: Los aislados de VIH-1 de 2 de 39 pacientes que recibieron terapia de combinación hasta por 2 años con zidovudina y didanosina mostraron una disminución de la sensibilidad a la zidovudina, didanosina, zalcitabina, estavudina y lamivudina en cultivo de célula. Estos aislados portaban cinco mutaciones (A62V, V75I, F77L, F116Y, y Q151M) en el gen de la transcriptasa reversa. En datos de estudios clínicos, se ha mostrado que la presencia de las mutaciones análogas a la timidina (M41L, D67N, L210W, T215Y, K219Q) reduce la respuesta a la didanosina. Farmacología: Toxicología Animal: Evidencia de toxicidad al músculo esquelético limitante de la dosis ha sido observada en ratones y ratas (pero no en perros) después de la dosificación a largo plazo (mayor que 90 días) con didanosina en dosis que eran aproximadamente 1,2 a 12 veces la exposición humana estimada. La relación de este hallazgo con el potencial de didanosina para causar miopatía en humanos no está clara. Sin embargo, la miopatía humana ha sido asociada con la administración de didanosina y otros análogos de nucleósidos. Farmacocinética: La didanosina es rápidamente absorbida, observándose las concentraciones plasmáticas pico generalmente de 0,25 a 1,50 horas después de la dosificación oral con una formulación tamponada. Los aumentos en las concentraciones plasmáticas de la didanosina fueron proporcionales a la dosis en el rango de 50-400 mg. Los parámetros farmacocinéticos al estado estable no difirieron significativamente de los valores obtenidos después de una dosis única. La unión de la didanosina a las proteínas plasmáticas in vitro fue baja ( < 5%). Basado en los datos de los estudios in vitro y en animales, se presume que el metabolismo de la didanosina en el hombre ocurre a través de las mismas vías responsables por la eliminación de las purinas endógenas. Comparación de las Formulaciones de Didanosina: En VIDEX® EC, el ingrediente activo, la didanosina, está protegida contra la degradación por el ácido del estómago mediante el uso de una cubierta entérica sobre los gránulos de la cápsula. La cubierta entérica se disuelve cuando los gránulos pasan al intestino delgado, el sitio de absorción del fármaco. Con las formulaciones tamponadas de didanosina, la administración con antiácidos provee protección de la degradación por el ácido del estómago. En voluntarios sanos, así como también en sujetos infectados con VIH-1, el área bajo la curva de concentración plasmática-tiempo (ABC) es equivalente para la didanosina administrada como las formulaciones VIDEX® EC en relación a una formulación de tableta tamponada. La concentración plasmática pico (Cmáx) de didanosina, administrada como VIDEX® EC, es reducida aproximadamente en 40% en relación con las tabletas tamponadas de didanosina. El tiempo para la concentración pico (Tmáx) aumenta desde aproximadamente 0,67 horas para las tabletas tamponadas de didanosina a 2,0 horas para VIDEX® EC. Efecto de la Comida sobre la Absorción de la Didanosina: En presencia de comida, la Cmáx y el ABC para VIDEX® EC fueron reducidas aproximadamente en 46% y 19%, respectivamente, en comparación con el estado de ayuno. VIDEX® EC debe ser tomado con el estómago vacío. Poblaciones Especiales: Insuficiencia Renal: Se recomienda que la dosis de VIDEX® EC sea modificada en los pacientes con disminución de la depuración de creatinina y en pacientes que reciban hemodiálisis de mantenimiento. Los datos de dos estudios utilizando una formulación tamponada de didanosina indicaron que la depuración oral aparente de la didanosina disminuyó y la vida media de eliminación terminal aumentó según disminuía la depuración de creatinina. Después de la administración oral, la didanosina no fue detectable en el fluido de diálisis peritoneal (n=6); la recuperación en el hemodializado (n=5) estaba en un rango de 0,6% a 7,4% de la dosis en un rango de un período de diálisis de 3-4 horas. La biodisponibilidad absoluta de la didanosina no fue afectada en pacientes que requerían diálisis. Pacientes Pediátricos: La farmacocinética de la didanosina administrada como VIDEX® EC no ha sido evaluada en pacientes pediátricos. Pacientes Geriátricos: La farmacocinética de la didanosina no ha sido estudiada en pacientes de más de 65 años de edad. Género: Los efectos del género sobre la farmacocinética de la didanosina no han sido estudiados.
Indicaciones.
VIDEX® EC, en combinación con otros agentes antirretrovirales, está indicado en el tratamiento de la infección por VIH-1 en adultos.
Dosificación.
Adultos: VIDEX® EC debe ser administrado con el estómago vacío. VIDEX® EC Cápsulas con Gránulos Recubiertos de Liberación Retardada deben ser tragadas intactas. La dosis diaria recomendada depende del peso corporal y se administra como una cápsula en un esquema de una vez al día como se muestra en la Tabla.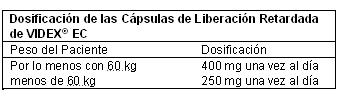
Pacientes Pediátricos: VIDEX® EC no ha sido estudiado en pacientes pediátricos. Por favor, consulte la información de prescripción completa para VIDEX® Polvo Pediátrico para Solución Oral para la dosificación y administración de didanosina a pacientes pediátricos. Ajuste de Dosis: Terapia Concomitante: En los pacientes que también están tomando tenofovir disoproxil fumarato, se recomienda una reducción de dosis de VIDEX® EC a 250 mg (adultos con un peso de por lo menos 60 kg con depuración de creatinina de por lo menos 60 mL/min) ó 200 mg (adultos con un peso menos que 60 kg con depuración de creatinina de por lo menos 60 mL/min) una vez al día, tomado conjuntamente con tenofovir y una comida ligera (400 kcalorías o menos y 20% de grasa o menos), o en estado de ayuno. No se ha establecido la dosis apropiada de VIDEX® EC administrado conjuntamente con tenofovir en pacientes con depuración de creatinina menor a 60 mL/min. Alteración de la Función Renal: Las recomendaciones de dosis para VIDEX® EC y VIDEX® Polvo Pediátrico para Solución Oral son diferentes para los pacientes con alteración de la función renal. Por favor, consulte la información sobre prescripción completa sobre la administración de VIDEX® Polvo Pediátrico para Solución Oral a pacientes con alteración de la función renal. En pacientes adultos con alteración de la función renal, la dosis de VIDEX® EC debiera ser ajustada para compensar por la tasa más lenta de eliminación. Las dosis y los intervalos de dosificación recomendados de VIDEX® EC en pacientes adultos con insuficiencia renal son presentados en la Tabla.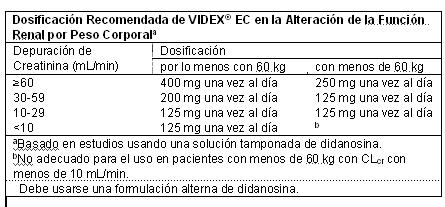
Pacientes que requieren Diálisis Peritoneal Ambulatoria Continua (DPAC) o Hemodiálisis: Para los pacientes que requieren DPAC o hemodiálisis, siga las recomendaciones de dosificación para los pacientes con depuración de creatinina menor que 10 mL/min, mostradas en la Tabla 13. No es necesario administrar una dosis suplementaria de didanosina después de la hemodiálisis.
Contraindicaciones.
VIDEX® EC está contraindicado en pacientes con hipersensibilidad clínicamente significativa previamente demostrada a cualquiera de los componentes de la formulación. Estas recomendaciones se basan en estudios de interacción medicamentosa o en toxicidades clínicas observadas. Alopurinol: La administración concomitante de didanosina y alopurinol está contraindicada debido a que se aumentan las exposiciones sistémicas de didanosina, lo que podría aumentar la toxicidad asociada con la didanosina. Ribavirina: La administración concomitante de didanosina y ribavirina está contraindicada debido a que aumentan las exposiciones del metabolito activo de la didanosina (dideoxiadenosina 5'-trifosfato). Han sido reportados en pacientes tratados conjuntamente con didanosina y ribavirina insuficiencia hepática fatal, así como neuropatía periférica, pancreatitis e hiperlactatemia sintomática/acidosis láctica.
Reacciones adversas.
Una toxicidad seria de la didanosina es la pancreatitis, la cual puede ser fatal. Otros efectos tóxicos importantes incluyen acidosis láctica/hepatomegalia severa con esteatosis; cambios retinianos y neuritis óptica; y neuropatía periférica. Cuando la didanosina es usada en combinación con otros agentes de toxicidad similar, la incidencia de toxicidad puede ser mayor que cuando la didanosina es usada sola. Así, los pacientes tratados con VIDEX® EC en combinación con estavudina, con o sin hidroxiurea, pueden tener un mayor riesgo de pancreatitis y hepatotoxicidad, las cuales pueden ser fatales, y de neuropatía periférica severa. Eventos adversos clínicos seleccionados que ocurrieron en un estudio de VIDEX® EC en combinación con otros agentes antirretrovirales son presentados en la Tabla.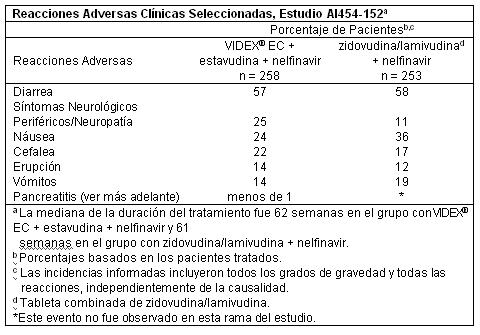
En estudios clínicos usando una formulación tamponada de didanosina, la pancreatitis que resultó en muerte, fue observada en un paciente que recibió didanosina más estavudina más nelfinavir, en un paciente que recibió didanosina más estavudina más indinavir, y en 2 de 68 pacientes que recibieron didanosina más estavudina más indinavir más hidroxiurea. En un programa de acceso temprano, la pancreatitis que resultó en muerte fue observada en un paciente que recibió VIDEX® EC más estavudina más hidroxiurea más ritonavir más indinavir más efavirenz. La frecuencia de pancreatitis está relacionada con la dosis. En estudios de Fase 3 usando unas formulaciones tamponadas de didanosina, la incidencia estaba en un rango de 1% a 10% con dosis mayores que las usualmente recomendadas y de 1% a 7% con la dosis recomendada. Observados Durante la Práctica Clínica: Los siguientes eventos han sido identificadas durante el uso después de la aprobación de formulaciones tamponadas de didanosina. Debido a que ellos son reportados voluntariamente por una población de tamaño desconocido, no pueden hacerse estimados de la frecuencia. Se han escogido estos eventos para su inclusión debido a su seriedad, la frecuencia de los informes reportados, la conexión causal con didanosina, o una combinación de esos factores. Cuerpo como un todo - dolor abdominal, alopecia, reacciones anafilactoides, astenia, escalofríos/fiebre, dolor y redistribución/acumulación de la grasa corporal. Trastornos Digestivos - anorexia, dispepsia y flatulencia. Trastornos de Glándulas Exocrinas - pancreatitis (incluyendo casos fatales), sialoadenitis, agrandamiento de las glándulas parótidas, boca seca y ojos secos. Trastornos Hematológicos - anemia, leucopenia y trombocitopenia. Hígado - hiperlactatemia sintomática/acidosis láctica y esteatosis hepática; hipertensión portal no cirrótica; hepatitis e insuficiencia hepática. Trastornos Metabólicos - diabetes mellitus, nivel elevado de la fosfatasa alcalina sérica, nivel elevado de la amilasa sérica, nivel elevado de la gamma-glutamil transferasa sérica, nivel elevado de ácido úrico sérico, hipoglucemia e hiperglucemia. Trastornos Musculoesqueléticos - mialgia (con o sin aumentos en la fosfokinasa de creatina), rabdomiólisis incluyendo insuficiencia renal aguda y hemodiálisis, artralgia y miopatía. Trastornos Oftalmológicos - despigmentación retiniana y neuritis óptica. Uso con Estavudina y Regímenes Basados en Hidroxiurea: Cuando la didanosina es usada en combinación con otros agentes de toxicidad similar, la incidencia de toxicidad puede ser mayor que cuando la didanosina es usada como monoterapia. Así, los pacientes tratados con VIDEX® EC en combinación con estavudina, con o sin hidroxiurea, pueden tener un mayor riesgo de padecer pancreatitis y hepatotoxicidad, las cuales pueden ser fatales, y de neuropatía periférica severa. Debe evitarse la combinación de VIDEX® EC e hidroxiurea, con o sin estavudina. Advertencias: Pancreatitis: pancreatitis fatal y no fatal han ocurrido durante la terapia con didanosina usada sola o en regímenes de combinación tanto en pacientes sin experiencia con el tratamiento o con experiencia con el tratamiento, independientemente del grado de inmunosupresión. VIDEX® EC deberá ser suspendido en pacientes con signos o síntomas de pancreatitis y descontinuado en pacientes con pancreatitis confirmada. Los pacientes tratados con VIDEX® EC en combinación con estavudina, con o sin hidroxiurea, pueden estar en riesgo de pancreatitis. Cuando se requiere tratamiento con medicamentos para el sostenimiento de la vida conocidos por causar toxicidad pancreática, se recomienda la suspensión de la terapia con VIDEX® EC. En los pacientes con factores de riesgo para pancreatitis, VIDEX® EC debiera ser usado con extrema precaución y solamente si está claramente indicado. Los pacientes con infección avanzada por VIH-1, especialmente los ancianos, tienen un riesgo aumentado de pancreatitis y debieran ser seguidos muy de cerca. Los pacientes con alteración de la función renal pueden estar en mayor riesgo de pancreatitis si son tratados sin ajuste de dosis. La frecuencia de la pancreatitis está relacionada con la dosis. En estudios de Fase 3 con formulaciones tamponadas de didanosina, la incidencia estaba en un rango de 1% a 10% con dosis mayores que aquellas recomendadas actualmente y de 1% a 7% con la dosis recomendada. Acidosis Láctica/Hepatomegalia con Esteatosis: Acidosis láctica y hepatomegalia severa con esteatosis, incluyendo casos fatales, han sido reportadas con el uso de análogos de nucleósidos solos o en combinación, incluyendo didanosina y otros antirretrovirales. Una mayoría de estos casos han sucedido en mujeres. La obesidad y la exposición prolongada a los nucleósidos pueden ser factores de riesgo. Se ha reportado acidosis láctica fatal en mujeres embarazadas que recibieron la combinación de didanosina y estavudina con otros agentes antirretrovirales. La combinación de didanosina y estavudina debe ser usada con precaución durante el embarazo y se recomienda solamente si el beneficio potencial supera, claramente, el riesgo potencial. Debe tomarse especial precaución cuando se administra VIDEX® EC a cualquier paciente con factores de riesgo conocidos para la enfermedad hepática; sin embargo, también se han reportado casos en pacientes sin factores de riesgo conocidos. El tratamiento con VIDEX® EC debiera ser suspendido en cualquier paciente que desarrolle hallazgos clínicos o de laboratorio sugestivos de hiperlactatemia sintomática, acidosis láctica o de hepatotoxicidad pronunciada (la cual puede incluir hepatomegalia y esteatosis aún en ausencia de elevaciones marcadas de las transaminasas). Cambios Retinianos y Neuritis Óptica: Cambios retinianos y neuritis óptica han sido reportados en pacientes que estaban tomando la didanosina. Exámenes periódicos de la retina debieran ser considerados para pacientes que reciben VIDEX® EC. Alteración Hepática y Toxicidad: Se desconoce si la alteración hepática afecta significativamente la farmacocinética de la didanosina. Por lo tanto, estos pacientes deben ser controlados de cerca por evidencia de toxicidad de la didanosina. La seguridad y la eficacia de VIDEX® EC no han sido establecidas en pacientes infectados con VIH con una significativa enfermedad hepática de base. Durante la terapia antirretroviral de combinación, los pacientes con disfunción hepática preexistente, incluyendo hepatitis activa crónica, tienen un aumento en la frecuencia de anormalidades de la función hepática, incluyendo eventos adversos hepáticos severos y potencialmente fatales, y deben ser controlados de acuerdo a la práctica estándar. Si hay evidencia de empeoramiento de la enfermedad hepática en tales pacientes, debe considerarse la interrupción o la descontinuación del tratamiento. La hepatotoxicidad y la insuficiencia hepática resultantes en la muerte fueron informadas durante la vigilancia post-mercadeo en pacientes infectados con VIH tratados con hidroxiurea y otros agentes antirretrovirales. Eventos hepáticos fatales fueron reportados más a menudo en pacientes tratados con la combinación de hidroxiurea, didanosina y estavudina. Esta combinación debe ser evitada. Hipertensión Portal No Cirrótica: Se han registrado casos de hipertensión portal no cirrótica en la etapa postcomercialización, entre los que se incluyen casos que condujeron a trasplante hepático o a la muerte. Se han confirmado casos de hipertensión portal no cirrótica asociada a la administración de didanosina, mediante biopsia hepática en pacientes sin evidencia de hepatitis viral. La aparición de los signos y síntomas osciló entre meses y años tras iniciar el tratamiento con didanosina. Las características comunes que se presentaron fueron aumento de las enzimas hepáticas, várices esofágicas, hematemesis, ascitis y esplenomegalia. Se debe monitorear a los pacientes que reciben VIDEX® EC a fin de detectar de manera temprana signos de hipertensión portal (por ejemplo, trombocitopenia y esplenomegalia) durante las visitas médicas de rutina. Se deben considerar análisis de laboratorio adecuados que incluyan las enzimas hepáticas, la bilirrubina sérica, la albúmina, el recuento sanguíneo completo y el índice normalizado internacional (INR) y la ultrasonografía. Se debe discontinuar la administración de VIDEX® EC en pacientes con indicios de hipertensión portal no cirrótica. Precauciones: Dosificación: VIDEX® EC debe ser administrado una vez al día con el estómago vacío. Neuropatía Periférica: La neuropatía periférica, manifestada por adormecimiento, hormigueo, o dolor en las manos o en los pies, ha sido reportada en pacientes que reciben terapia con didanosina. La neuropatía periférica ha ocurrido más frecuentemente en pacientes con enfermedad avanzada por VIH, en pacientes con una historia de neuropatía, o en pacientes tratados con terapia con medicamentos neurotóxicos, incluyendo estavudina. Se debe considerar la suspensión de VIDEX® EC en los pacientes que desarrollen neuropatía periférica. Redistribución de la Grasa Corporal: Redistribución/acumulación de la grasa corporal, incluyendo la obesidad central, aumento de la grasa dorsocervical (giba de búfalo), adelgazamiento periférico, adelgazamiento facial, aumento de tamaño de las mamas, y la "apariencia cushingoide" han sido observados en pacientes que reciben terapia antirretroviral. El mecanismo y las consecuencias a largo plazo de estos eventos, no se conocen actualmente. No se ha establecido una relación causal. Síndrome de Reconstitución Inmune: El síndrome de reconstitución inmune ha sido reportado en pacientes tratados con la terapia antirretroviral de combinación, incluyendo VIDEX® EC. Durante la fase inicial del tratamiento antirretroviral de combinación, los pacientes cuyo sistema inmune responde pueden desarrollar una respuesta inflamatoria a infecciones oportunísticas indolentes o residuales (tales como la infección por Mycobacterium avium, citomegalovirus, neumonía por Pneumocystis jiroveci [PCP], o tuberculosis), las cuales pueden requerir evaluación y tratamiento adicionales. También se ha informado acerca de la aparición de enfermedades autoinmunes (tales como enfermedad de Graves, polimiositis y síndrome de Guillain-Barré) en el contexto de la reconstitución inmunológica; sin embargo, el tiempo hasta el inicio es más variable, y dichas enfermedades se pueden producir varios meses después de iniciado el tratamiento. General: Pacientes con Alteración de la Función Renal: Los pacientes con alteración de la función renal (depuración de creatinina < 60 mL/min) pueden estar en mayor riesgo de toxicidad por didanosina debido a una disminución en la depuración del fármaco. Se recomienda una reducción de la dosis en estos pacientes. Carcinogénesis y Mutagénesis: Los estudios de carcinogenicidad de por vida fueron conducidos en ratones y ratas por 22 y 24 meses, respectivamente. En el estudio en el ratón, dosis iniciales de 120, 800 y 1200 mg/kg/día para cada sexo fueron disminuidas después de 8 meses a 120, 210 y 210 mg/kg/día para las hembras y 120, 300 y 600 mg/kg/día para los machos. Las dos dosis mayores excedían la dosis máxima tolerada en los machos. La dosis baja en las hembras representaba 0,68 veces la exposición máxima en humanos y la dosis intermedia en los machos representaba 1,7 veces la exposición máxima en humanos basado en comparaciones relativas del ABC. En el estudio en la rata, las dosis iniciales eran 100, 250 y 1000 mg/kg/día, y la dosis más alta fue disminuida a 500 mg/kg/día después de 18 meses. La dosis superior en las ratas macho y hembra representaba 3 veces la exposición máxima en humanos. La didanosina no indujo aumentos significativos en las lesiones neoplásicas en las ratas y los ratones a las dosis máximas toleradas. La didanosina fue positiva en los siguientes ensayos de toxicología genética: 1) ensayo de mutagenicidad bacteriana en la cepa de prueba de Escherichia coli WP2 uvrA; 2) ensayo de mutación de genes de células de mamíferos de L5178Y/TK+/-células de linfoma de ratón; 3) ensayo in vitro de aberraciones cromosomales en linfocitos periféricos humanos cultivados; 4) ensayo de aberraciones cromosomales in vitro en células de Pulmón de Hamster Chino; y 5) ensayo de transformación in vitro de BALB/c 3T3. No se observó evidencia de mutagenicidad en el ensayo de mutagenicidad bacteriana de Ames en Salmonella o en el ensayo de micronúcleos de rata y de ratón in vivo. Gestación, Reproducción y Fertilidad: Categoría B de la Gestación. Se han realizado estudios de reproducción en ratas y conejos en dosis hasta 12 y 14,2 veces la exposición estimada en humanos (basados en los niveles plasmáticos), respectivamente, y no han revelado evidencia de alteración de la fertilidad o daño al feto debido a la didanosina. A aproximadamente 12 veces la exposición estimada en humanos, la didanosina fue ligeramente tóxica en las ratas hembra y sus crías durante la lactancia media y tardía. Estas ratas mostraron una reducción en la ingesta de alimento y ganancia de peso corporal pero el desarrollo físico y funcional de las crías no fue alterado y no hubo mayores cambios en la generación F2. Un estudio en ratas demostró que la didanosina y/o sus metabolitos son transferidos al feto a través de la placenta. Los estudios sobre la reproducción en animales no siempre son predictivos de la respuesta en humanos. No hay estudios adecuados y bien controlados de didanosina en mujeres embarazadas. La didanosina debe ser usada durante el embarazo solamente si el beneficio potencial justifica el riesgo potencial. Se ha reportado acidosis láctica fatal en mujeres embarazadas que recibieron la combinación de didanosina y estavudina con otros agentes antirretrovirales. No está claro si el embarazo aumenta el riesgo del síndrome de acidosis láctica/esteatosis hepática reportado en mujeres no embarazadas que reciben análogos de nucleósidos (ver Advertencias: Acidosis Láctica/Hepatomegalia Severa con Esteatosis). La combinación de didanosina y estavudina debe ser usada con precaución durante el embarazo y se recomienda solamente si el beneficio potencial supera, claramente, el riesgo potencial. Los profesionales de la salud que atienden mujeres embarazadas infectadas con VIH que reciben didanosina, deben permanecer en alerta para un diagnóstico temprano del síndrome de acidosis láctica/esteatosis hepática. Madres Lactantes: Los Centros para el Control y Prevención de Enfermedades del Servicio de Salud Pública de los Estados Unidos aconsejan a las mujeres infectadas por VIH no amamantar sus infantes para evitar el riesgo de la transmisión postnatal de VIH. Un estudio en ratas demostró que después de la administración oral, la didanosina y/o sus metabolitos fueron excretados en la leche de las ratas lactantes. No se conoce si la didanosina es excretada en la leche humana. Debido al potencial para la transmisión del VIH y el potencial para reacciones adversas serias en lactantes, se debe instruir a las madres para no amamantar si están recibiendo VIDEX® EC. Uso Pediátrico: La seguridad y la eficacia de VIDEX® EC en pacientes pediátricos no han sido establecidas. Por favor, consulte la información de prescripción completa para VIDEX® Polvo Pediátrico para Solución Oral para la dosificación y administración de didanosina a pacientes pediátricos. Uso Geriátrico: En un Programa de Acceso Expandido usando una formulación tamponada de didanosina para el tratamiento de la infección avanzada por VIH, los pacientes con 65 años de edad o más tenían una mayor frecuencia de pancreatitis (10%) que los pacientes jóvenes (5%). Estudios clínicos de didanosina, incluyendo aquellos para VIDEX® EC, no incluyeron suficiente número de sujetos de 65 años o más para determinar si ellos responden en forma diferente a los sujetos más jóvenes. Se sabe que la didanosina es sustancialmente excretada por los riñones, y el riesgo de reacciones tóxicas a este medicamento puede ser mayor en pacientes con alteración de la función renal. Debido a que los pacientes ancianos tienen mayor probabilidad de una función renal reducida, debe tenerse cuidado en la selección de la dosis. Además, la función renal debe ser controlada y deben hacerse ajustes de dosis de acuerdo con ello.
Interacciones.
Las recomendaciones clínicas basadas en los resultados de estos estudios son presentadas en la Tabla.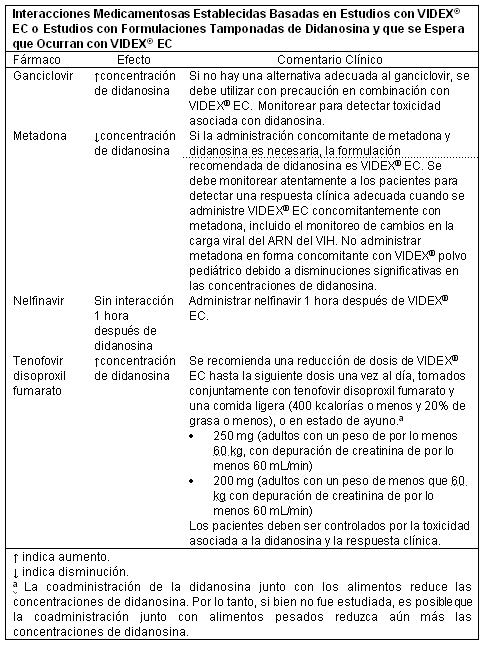
La coadministración de VIDEX® EC con medicamentos que se sabe causan pancreatitis, puede aumentar el riesgo de esta toxicidad: Las interacciones medicamentosas predichas con VIDEX® EC son presentadas en la Tabla.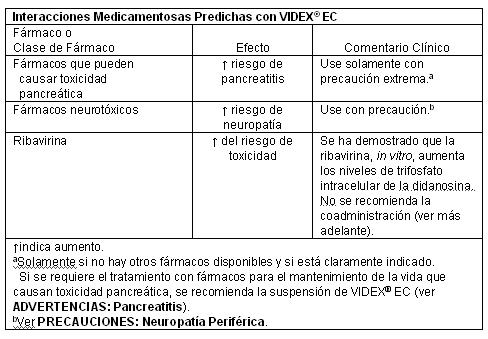
Análogos de Nucleósido/Nucleótido: Tenofovir disoproxil fumarato. La exposición a la didanosina aumenta cuando es coadministrada con tenofovir (ver las Tablas 3, 5 y 8). El aumento de la exposición puede causar o empeorar la toxicidad clínica relacionada con la didanosina, incluyendo pancreatitis, hiperlactatemia sintomática/acidosis láctica, y neuropatía periférica. La administración simultánea de tenofovir con VIDEX® EC se debe realizar con precaución, y los pacientes deben ser controlados cuidadosamente por la toxicidad relacionada a la didanosina y la respuesta clínica. Se debe suspender el VIDEX® EC si se desarrollan signos o síntomas de pancreatitis, hiperlactatemia sintomática, o acidosis láctica. La administración de dosis reducidas de VIDEX® EC con tenofovir y una comida ligera resultó en exposiciones de didanosina (ABC) similares a las dosis recomendadas de VIDEX® EC administrado solo, en estado de ayuno (ver la Tabla 3). Por lo tanto, cuando se administra con tenofovir, se recomienda una reducción de dosis de VIDEX® EC a 250 mg (adultos con un peso de por lo menos 60 kg, con depuración de creatinina de por lo menos 60 mL/min) ó 200 mg (adultos con un peso menos que 60 kg con depuración de creatinina de por lo menos 60 mL/min) una vez al día, y ambas drogas se pueden tomar juntas con una comida ligera (400 kcalorías o menos y 20% de grasa o menos), o en estado de ayuno. La coadministración de didanosina con alimento disminuye las concentraciones de didanosina. Por eso, aunque no se ha estudiado, es posible que la coadministración con comidas más pesadas pudiera reducir aún más las concentraciones de didanosina. Ribavirina: La exposición al metabolito activo de la didanosina (dideoxiadenosina 5'-trifosfato) aumenta cuando la didanosina es coadministrada con ribavirina (ver la Tabla 9). Se ha reportado insuficiencia hepática fatal, así como también neuropatía periférica, pancreatitis e hiperlactatemia sintomática/acidosis láctica en pacientes que reciben tanto didanosina como ribavirina. No se recomienda la coadministración de didanosina y ribavirina.
Conservación.
Las Cápsulas de Liberación Retardada de VIDEX® EC deben ser almacenadas en frascos herméticamente cerrados. Conserve a temperatura ambiente no mayor a 30°C. Manejo y disposición: Las opciones de disposición incluyen incineración, tierras rehabilitadas, o alcantarillas según sea dictado por circunstancias específicas y reglamentos nacionales, estatales y locales relevantes.
Sobredosificación.
No hay antídoto conocido para la sobredosificación con didanosina. En estudios de Fase 1, en los cuales formulaciones tamponadas de didanosina fueron inicialmente administradas en dosis diez veces la dosis recomendada actualmente, los efectos tóxicos incluyeron: pancreatitis, neuropatía periférica, diarrea, hiperuricemia y disfunción hepática. La didanosina no es dializable por diálisis peritoneal, aunque hay cierta depuración por hemodiálisis.
Presentación.
VIDEX® EC (didanosina) Cápsulas con Gránulos Recubiertos de Liberación Retardada: son cápsulas blancas, opacas, que están empacadas en frasco con tapa resistente a los niños como se describe en la Tabla. Cápsulas 250 mg: envase con 30 cápsulas de acción prolongada en formulación de microgránulos. Cápsulas 400 mg: envase con 30 cápsulas de acción prolongada en formulación de microgránulos.