XELJANZ®
PFIZER
Antirreumático.
Composición.
Cada comprimido recubierto contiene 8,078 mg de citrato de tofacitinib equivalente a 5 mg de base libre del principio activo de tofacitinib. Excipientes: Celulosa microcristalina, Lactosa Monohidrato, Croscarmelosa sódica, Estereato de magnesio, HPMC 2910/Hipromelosa 6cP, Dióxido de titanio, Macrogol/PEG3350, Triacetina (triacetato de glicerol).
Toxicología.
En estudios no clínicos, se observaron efectos en el sistema inmunitario y hematopoyético, que se atribuyeron a las propiedades farmacológicas (inhibición JAK) de tofacitinib. Se observaron efectos secundarios de la inmunosupresión, por ejemplo, infecciones virales y bacterianas y linfoma, con dosis clínicamente relevantes. Otros resultados con dosis muy superiores a las exposiciones en seres humanos incluyeron efectos en los sistemas hepático y gastrointestinal. Se observó linfoma en 3 de 8 monos adultos y en 0 de 14 monos jóvenes que recibieron dosis de tofacitinib de 5 mg/kg BID. El nivel sin efectos adversos observados (NOAEL) para los linfomas fue de 1 mg/kg BID. El AUC no ligado con 1 mg/kg BID fue de 341 ng•h/mL, que es aproximadamente la mitad del AUC no ligado con 10 mg BID y similar al AUC no ligado con 5 mg BID en seres humanos. Tofacitinib no es mutagénico ni genotóxico sobre la base de los resultados de una serie de pruebas in vitro e in vivo para determinar mutaciones genéticas y aberraciones cromosómicas. El potencial carcinógeno de tofacitinib se evaluó en estudios de carcinogenia en ratones transgénicos rasH2 de 6 meses y en estudios de carcinogenia en ratas de 2 años. Tofacitinib no fue carcinógeno en los ratones con una dosis alta de hasta 200 mg/kg/día (AUC del fármaco no ligado de ~ 19 veces el AUC en seres humanos con 10 mg BID). Se observaron tumores benignos de células de Leydig en las ratas: los tumores benignos de células de Leydig en las ratas no están asociados con un riesgo de tumores de células de Leydig en los seres humanos. Se observaron hibernomas (tumores malignos del tejido adiposo pardo) en las ratas hembra con dosis ≥ 30 mg/kg/día (AUC del fármaco no ligado de ~ 41 veces el AUC en seres humanos con 10 mg BID). Se observaron timomas benignos en las ratas hembra con dosis de solamente 100 mg/kg/día que se redujeron a 75 mg/kg/día (AUC del fármaco no ligado de ~ 94 veces el AUC en seres humanos con 10 mg BID). Se demostró que tofacitinib es teratógeno en ratas y conejos, y que tiene efectos en la fertilidad, el parto y el desarrollo peri/posnatal en las ratas hembra. Tofacitinib no tuvo efectos en la fertilidad de los machos ni en la concentración o la movilidad del esperma. Se encontró tofacitinib en la leche de ratas lactantes.
Indicaciones.
Está indicado para el tratamiento de pacientes adultos con artritis reumatoide moderada a severamente activa, quienes han tenido una respuesta inadecuada o intolerancia a metotrexato. Puede ser usado como monoterapia o en combinación con metotrexato u otras drogas no biológicas modificadoras de la enfermedad (DMARDs).
Dosificación.
Posología: Xeljanz puede utilizarse como monoterapia o en combinación con metotrexato u otros DMARD no biológicos. La posología recomendada es de 5 mg administrados dos veces al día. El uso de Xeljanz no se ha estudiado y no debe usarse en combinación con DMARD biológicos, como antagonistas del TNF, antagonistas de IL-1R, antagonistas de IL-6R, anticuerpos monoclonales anti-CD20 y moduladores selectivos de la coestimulación, y con inmunosupresores potentes, como azatioprina, ciclosporina y tacrolimus, debido a la posibilidad de que aumente la inmunosupresión y el riesgo de infecciones. El tratamiento con Xeljanz debe interrumpirse si un paciente presenta una infección grave hasta lograr controlarla. Ajustes de dosis debido a resultados anormales en análisis clínicos: Puede ser necesario ajustar la dosis o interrumpir la administración de la dosis para tratar los resultados anormales relacionados con la dosis en los análisis clínicos, entre ellos, neutropenia, anemia y linfopenia, como se describe en la Tabla 1, Tabla 2 y Tabla 3, a continuación. Se recomienda no comenzar el tratamiento con Xeljanz en pacientes con un recuento absoluto de neutrófilos (RAN) < 1000/mm3, un conteo de linfocitos menor a 500 células/mm3 o quienes tienen niveles de hemoglubina menores de 9 g/dL.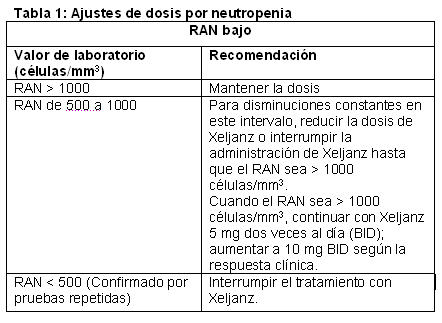
Se recomienda no comenzar el tratamiento con Xeljanz en pacientes con Hg < 9 g/dL.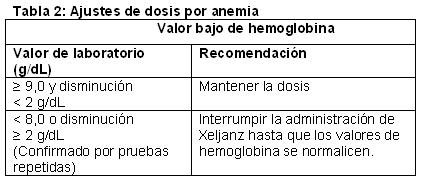
Poblaciones especiales: Insuficiencia renal: No es necesario ajustar la dosis en pacientes con insuficiencia renal leve. La dosis de Xeljanz no debe superar los 5 mg una vez al día en pacientes con insuficiencia renal grave moderada o grave. Insuficiencia hepática: Xeljanz no debe utilizarse en pacientes con insuficiencia hepática grave. La dosis de Xeljanz no debe superar los 5 mg una vez al día en pacientes con insuficiencia hepática moderada. Pacientes que reciben inhibidores del citocromo P450 (CYP3A4) y del citocromo 2C19: La dosis de Xeljanz no debe superar los 5 mg BID en pacientes que reciben inhibidores potentes del CYP3A4. La dosis de Xeljanz no debe superar los 5 mg BID en pacientes que reciben uno o más medicamentos concomitantes que causan la inhibición moderada del CYP3A4 y la inhibición potente del CYP2C19 (por ejemplo, fluconazol). La coadministración de Xeljanz con inductores potentes del CYP3A4 puede causar la pérdida o la reducción de la respuesta clínica. Pacientes ancianos (≥ 65 años): No es necesario ajustar la dosis a pacientes de 65 años o mayores. Población pediátrica: No se han establecido la seguridad y la eficacia de Xeljanz en los niños, desde neonatos hasta menores de 18 años. Método de administración: Xeljanz se administra por vía oral, con o sin alimentos.
Contraindicaciones.
Ninguna.
Reacciones adversas.
Los siguientes datos incluyen 5 estudios multicéntricos, controlados, doble ciego. En estos estudios, los pacientes se aleatorizaron para recibir dosis de Xeljanz 5 mg BID (243 pacientes) y 10 mg BID (245 pacientes) como monoterapia y de Xeljanz 5 mg BID (973 pacientes) y 10 mg BID (969 pacientes) en combinación con DMARD (incluso metotrexato). De los 3030 pacientes que recibieron Xeljanz en estos 5 estudios clínicos, 1871 recibieron tratamiento durante al menos 6 meses, y 580, durante al menos un año. La población de seguridad a largo plazo incluye a todos los pacientes que participaron en un estudio doble ciego, controlado (incluso los estudios anteriores en fase de desarrollo) y luego participaron en uno de dos estudios de seguridad a largo plazo. De los 3227 pacientes que recibieron Xeljanz en los estudios a largo plazo, 1689 recibieron tratamiento abierto durante al menos 6 meses; 970, durante al menos un año; 659 recibieron tratamiento durante al menos 2 años; y 62, durante 3 años. Todos los pacientes de estos estudios tenían artritis reumatoide activa de moderada a grave. La población del estudio tenía una edad promedio de 54 años, y el 84% eran mujeres. Experiencia en estudios clínicos: Las reacciones adversas graves más frecuentes fueron las infecciones grave. Las reacciones adversas informadas con mayor frecuencia durante los primeros 3 meses en los estudios clínicos controlados (que se produjeron en ≥ 2% de los pacientes tratados con Xeljanz como monoterapia o en combinación con DMARD) fueron infecciones de las vías respiratorias superiores, dolor de cabeza, nasofaringitis y diarrea. La proporción de pacientes que interrumpieron el tratamiento debido a cualquier reacción adversa durante los estudios doble ciego, controlados con placebo, fue del 6,8% para los pacientes que tomaban Xeljanz y del 3,7% para los pacientes tratados con placebo. Las reacciones adversas más frecuentes que causaron la interrupción de Xeljanz fueron las infecciones. Las infecciones más frecuentes que llevaron a la interrupción del tratamiento fueron el herpes zóster y la neumonía. Las reacciones adversas al fármaco que se mencionan en la siguiente tabla se presentan por categoría de sistema corporal y frecuencia, definida mediante esta convención: muy frecuente (≥ 1/10), frecuente (≥ 1/100 a < 1/10), infrecuente (≥ 1/1000 a < 1/100) o raro (≥ 1/10 000 a < 1/1000). Para cada grupo de frecuencia, los efectos indeseables se presentan en orden decreciente de gravedad.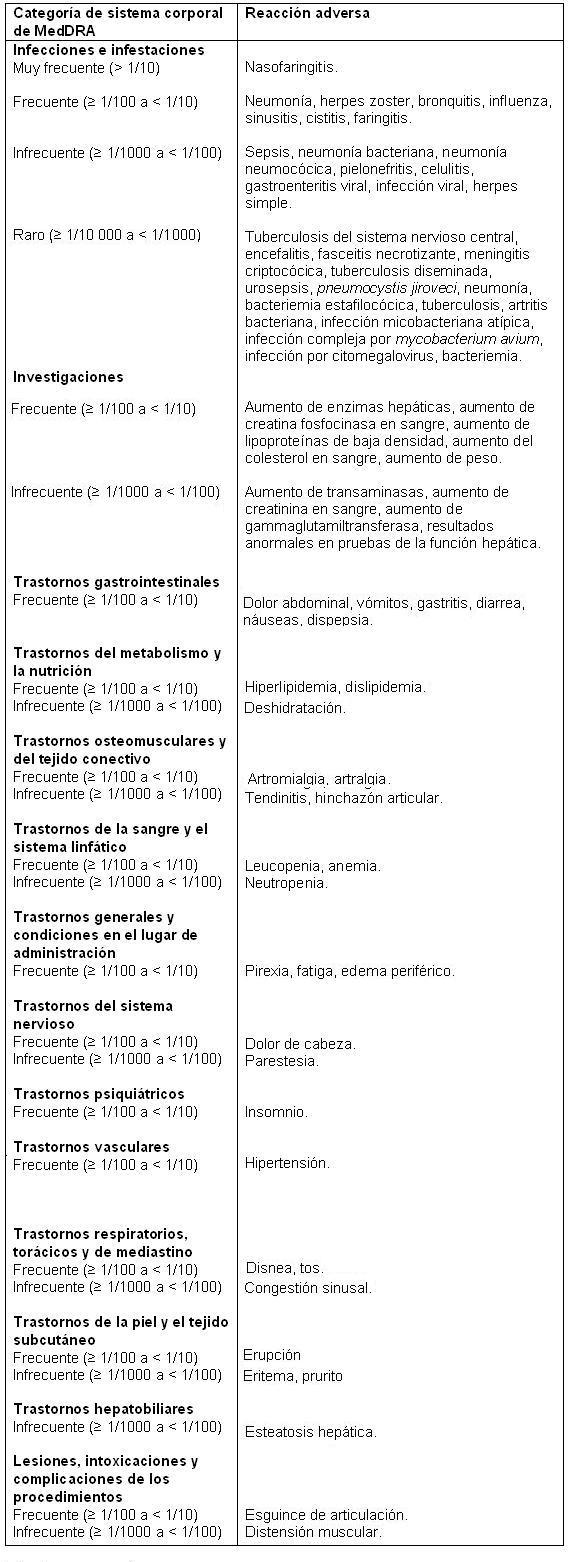
Infecciones generales: En el estudio clínico controlado de 6 meses, la tasa de infecciones en el grupo de monoterapia con 5 mg BID y 10 mg BID de Xeljanz fue del 16,5% y el 19,2%, respectivamente, en comparación con el 18,9% en el grupo PBO. En estudios con una duración de 6 o 12 meses sin tratamiento de fondo con DMARD, la tasa de infecciones en el grupo de 5 mg BID y 10 mg BID de Xeljanz más DMARD fue del 20,9% y el 21,7%, respectivamente, en comparación con el 18,2% en el grupo placebo más DMARD. Las infecciones informadas con mayor frecuencia fueron las infecciones de las vías respiratorias superiores y la nasofaringitis (4,1% y 3,4%, respectivamente). La tasa global de infecciones con Xeljanz en la población de exposición completa de seguridad a largo plazo fue de 41,5 eventos por 100 años-paciente (31,5 y 66,9 para 5 mg y 10 mg BID, respectivamente). Para los pacientes con monoterapia, la tasa fue de 35,5 y 55,8 eventos por 100 años-paciente para 5 mg y 10 mg BID, respectivamente. Para los pacientes con tratamiento de fondo con DMARD, la tasa fue de 28,8 y de 78,4 eventos por 100 años paciente para 5 mg y 10 mg BID, respectivamente. En el estudio clínico controlado de 6 meses, la tasa de infecciones graves en el grupo de monoterapia con 5 mg BID de Xeljanz fue de 0,85 eventos por 100 años-paciente. En el grupo de monoterapia con 10 mg BID de Xeljanz, fue de 3,5 eventos por 100 años-paciente, y fue de 0 eventos por 100 años-paciente en el grupo PBO. En los estudios con una duración de 6 o 12 meses, las tasas de infecciones graves en los grupos de 5 mg BID y 10 mg BID de Xeljanz más DMARD fueron de 3,6 y 2,9 eventos por 100 años-paciente, respectivamente, en comparación con 1,7 eventos por 100 años-paciente en el grupo placebo más DMARD. En la población de exposición completa de seguridad a largo plazo, la tasa global de infecciones graves fue de 2,3 eventos y de 4,9 eventos por 100 años-paciente para 5 mg y 10 mg BID de Xeljanz, respectivamente. Las infecciones graves más frecuentes informadas con Xeljanz incluyeron neumonía, infección de las vías urinarias y herpes zoster. Se han informado casos de infecciones oportunistas. De los 3315 pacientes que se inscribieron en los estudios I a V, 505 pacientes con artritis reumatoide tenían 65 años o más, incluso 71 pacientes tenían 75 años o más. La frecuencia de infecciones graves entre los sujetos de 65 años o más tratados con Xeljanz fue más alta que en aquellos menores de 65 años. Debido a que hay una mayor incidencia de infecciones en los ancianos en general, se debe tener precaución al tratar a esta población. Análisis de laboratorio: Neutrófilos: En los estudios clínicos controlados se confirmaron disminuciones del RAN por debajo de 1000/mm3 en 0,08% de los pacientes en los grupos de 5 mg BID y 10 mg BID de Xeljanz. No hubo disminuciones confirmadas del RAN por debajo de 500 por mm3 en ningún grupo de tratamiento. No hubo una relación clara entre la neutropenia y la aparición de infecciones graves. En la población de seguridad a largo plazo, el patrón y la incidencia de las disminuciones confirmadas en el RAN fueron coherentes con lo observado en los estudios clínicos controlados. Análisis de enzimas hepáticas: Con poca frecuencia, se observaron aumentos confirmados de las enzimas hepáticas > 3x el límite normal superior (ULN). En los pacientes que tuvieron un aumento de enzimas hepáticas, la modificación del tratamiento, por ejemplo, la reducción de la dosis del DMARD concomitante, la interrupción de Xeljanz o la reducción de la dosis de Xeljanz, generó la disminución o la normalización de las enzimas hepáticas. Estos aumentos no se asociaron con aumentos clínicamente relevantes de la bilirrubina directa ni con evidencia clínica de hepatitis o de insuficiencia hepática. En la parte controlada del estudio de monoterapia de fase 3 (de 0 a 3 meses), se observaron aumentos en las concentraciones de ALT > 3x ULN en 1,65%, 0,41% y 0% de los pacientes que recibieron placebo, 5 mg y 10 mg BID, respectivamente. En este estudio, se observaron aumentos en las concentraciones de AST > 3x ULN en 1,65%, 0,41% y 0% de los pacientes que recibieron placebo, 5 mg y 10 mg BID, respectivamente. En la parte controlada de los estudios de fase 3 con tratamiento de fondo con DMARD (de 0 a 3 meses), se observaron aumentos en las concentraciones de ALT > 3x ULN en 0,9%, 1,24% y 1,25% de los pacientes que recibieron placebo, 5 mg y 10 mg BID, respectivamente. En estos estudios, se observaron aumentos en las concentraciones de AST > 3x ULN en 0,72%, 0,5% y 0,42% de los pacientes que recibieron placebo, 5 mg y 10 mg BID, respectivamente. Lípidos: Los aumentos en los parámetros lipídicos (colesterol total, colesterol LDL, colesterol HDL, triglicéridos) se evaluaron por primera vez un mes después del inicio de Xeljanz en los estudios clínicos doble ciego, controlados. Se observaron aumentos en este punto temporal, que permanecieron estables posteriormente. A continuación se resumen los cambios en los parámetros lipídicos desde el momento basal hasta el final del estudio (6 o 12 meses) en los estudios clínicos controlados. El colesterol LDL medio aumentó 14% en el grupo de Xeljanz 5 mg BID y 20% en el grupo de Xeljanz 10 mg BID. El colesterol HDL medio aumentó 16% en el grupo de Xeljanz 5 mg BID y 18% en el grupo de Xeljanz 10 mg BID. Los cocientes del colesterol LDL/HDL medio, básicamente no se modificaron en los pacientes tratados con Xeljanz. Los cocientes de apolipoproteína B (apoB)/apoA1, básicamente no se modificaron en los pacientes tratados con Xeljanz. En un estudio clínico controlado, los aumentos en el colesterol LDL y la apoB disminuyeron a las concentraciones anteriores al tratamiento en respuesta al tratamiento con estatinas. En la población de seguridad a largo plazo, los aumentos en los parámetros lipídicos fueron coherentes con lo observado en los estudios clínicos controlados.
Advertencias.
Infecciones graves: Se han informado infecciones graves y a veces mortales debido a patógenos bacterianos, micobacterianos, micóticos invasivos y virales o a otros patógenos oportunistas en pacientes con artritis reumatoide que reciben agentes inmunomoduladores, entre ellos, DMARD y Xeljanz. Las infecciones graves más frecuentes informadas con Xeljanz incluyeron neumonía, celulitis, herpes zóster e infección de las vías urinarias. Entre las infecciones oportunistas, con el uso de Xeljanz se informaron tuberculosis y otras infecciones micobacterianas, Cryptococcus, candidiasis esofágica, herpes zóster multidermatomal, citomegalovirus y virus BK. Algunos pacientes han presentado la enfermedad diseminada, en lugar de localizada; estos pacientes a menudo tomaban agentes inmunomoduladores concomitantes, como metotrexato o corticosteroides, que, además de la artritis reumatoide, pueden predisponerlos a sufrir infecciones. También pueden ocurrir otras infecciones graves, que no se informaron en los estudios clínicos (por ejemplo, histoplasmosis, coccidioidomicosis y listeriosis). Xeljanz no debe administrarse a pacientes con infección activa, que incluye las infecciones localizadas. Se deben considerar los riesgos y los beneficios del tratamiento antes de comenzar a administrar Xeljanz a pacientes con infecciones crónicas o recurrentes, que hayan estado expuestos a tuberculosis, que tengan antecedentes de una infección grave u oportunista, que hayan vivido en zonas de tuberculosis endémica o micosis endémica o viajado a estas zonas, o que tengan afecciones subyacentes que puedan predisponerlos a las infecciones. Se debe supervisar cuidadosamente a los pacientes para detectar el desarrollo de signos y síntomas de infección durante el tratamiento con Xeljanz y después de él. El tratamiento con Xeljanz debe interrumpirse si un paciente presenta una infección grave, una infección oportunista o sepsis. Se deben hacer pruebas diagnósticas completas y de inmediato (adecuadas para un paciente inmunodeprimido), comenzar el tratamiento antimicrobiano adecuado y supervisar cuidosamente a un paciente que presente una infección nueva durante el tratamiento con Xeljanz. Debido a que hay una mayor incidencia de infecciones en los ancianos en general, se debe tener precaución al tratar a esta población. Tuberculosis: Se debe evaluar a los pacientes y determinar si tienen infección latente o activa antes de la administración de Xeljanz. También se debe considerar el tratamiento antituberculosis antes de la administración de Xeljanz a pacientes con antecedentes de tuberculosis latente o activa en los que no se puede confirmar un ciclo adecuado de tratamiento y a pacientes con resultados negativos en una prueba de tuberculosis latente que tienen factores de riesgo de infección por tuberculosis. Se recomienda consultar a un profesional de salud con experiencia en el tratamiento de la tuberculosis como ayuda para decidir si es adecuado comenzar el tratamiento antituberculosis en un paciente individual. Se debe supervisar cuidadosamente a los pacientes para detectar el desarrollo de signos y síntomas de tuberculosis, incluso a los pacientes que tuvieron resultados negativos en las pruebas de infección por tuberculosis latente antes de comenzar el tratamiento. La incidencia de tuberculosis en los programas de desarrollo clínico de Xeljanz en todo el mundo es del 0,1 % al 0,2 %. Los pacientes con tuberculosis latente deben recibir tratamiento antimicobacteriano estándar antes de la administración de Xeljanz. Reactivación viral: Se ha informado la reactivación viral con el tratamiento con DMARD, y se observaron casos de reactivación del virus del herpes (por ejemplo, herpes zoster) en estudio clínicos con Xeljanz. Se desconoce la repercusión de Xeljanz en la reactivación de la hepatitis viral crónica. Se excluyó de los estudios clínicos a los pacientes con resultados positivos de hepatitis B y C. Trastorno linfoproliferativo y neoplasia: Existe la posibilidad de que Xeljanz afecte las defensas del huésped contra las neoplasias. Se desconoce la repercusión del tratamiento con Xeljanz en el desarrollo y la evolución de las neoplasias, pero se observaron neoplasias en los estudios clínicos. En los estudios clínicos controlados, se diagnosticaron 13 neoplasias (sin incluir el cáncer de piel de tipo no melanoma [NMSC]) en los pacientes que recibieron Xeljanz, en comparación con 0 neoplasias (sin incluir el NMSC) en el grupo placebo/placebo más DMARD. Más de 3000 pacientes (2098 años-paciente de observación) recibieron tratamiento con Xeljanz durante hasta 1 año, mientras que aproximadamente 680 pacientes (203 años-paciente de observación) recibieron tratamiento con placebo durante un máximo de 6 meses. La tasa de incidencia ajustada según la exposición fue de 0,62 eventos por 100 años-paciente en los grupos de Xeljanz. Se han observado linfomas en pacientes tratados con Xeljanz. Si bien los pacientes con artritis reumatoide, en especial quienes tienen una enfermedad muy activa, pueden correr más riesgo (hasta varios múltiplos) de presentar linfoma, se desconoce la función de la inhibición de la cinasa asociada con Janus (JAK) en el desarrollo del linfoma. En la población de seguridad a largo plazo, la tasa de neoplasias (sin incluir el NMSC) fue de 1,12 eventos por 100 años-paciente, coherente con la tasa observada en el período controlado. Perforaciones gastrointestinales: Se han informado eventos de perforación gastrointestinal en estudios clínicos en pacientes con artritis reumatoide, aunque se desconoce la función de la inhibición JAK en estos eventos. La tasa de incidencia de perforación gastrointestinal entre todos los estudios (fase 2, fase 3 y extensión a largo plazo) fue de 0,177 eventos por 100 años-paciente con el tratamiento con Xeljanz. Los eventos se informaron principalmente como perforación diverticular, peritonitis, absceso abdominal y apendicitis. Todos los pacientes que tuvieron perforaciones gastrointestinales también estaban tomando antiinflamatorios no esteroideos (AINEs) o corticosteroides concomitantes. Se desconoce la contribución relativa de estos medicamentos concomitantes frente a Xeljanz en el desarrollo de las perforaciones gastrointestinales. Xeljanz debe usarse con precaución en los pacientes que pueden correr mayor riesgo de perforación gastrointestinal (por ejemplo, pacientes con antecedentes de diverticulitis). Los pacientes con síntomas abdominales nuevos deben evaluarse de inmediato para identificar de forma temprana la perforación gastrointestinal. Parámetros de laboratorio: Neutrófilos: El tratamiento con Xeljanz se asoció con una mayor incidencia de neutropenia ( < 2000/mm3) en comparación con placebo. No se recomienda comenzar el tratamiento con Xeljanz en pacientes con recuento bajo de neutrófilos (es decir, RAN < 1000/mm3). Para pacientes que tienen un RAN constante de 500 a 1000/mm3, se debe reducir la dosis de Xeljanz o interrumpir la administración de la dosis hasta que el RAN sea > 1000 células/mm3. No se recomienda el tratamiento en pacientes que presentan un recuento absoluto de neutrófilos < 500 por mm3. Se debe supervisar la concentración de neutrófilos después de 4 a 8 semanas de tratamiento y cada 3 meses posteriormente. Hemoglobina: No se recomienda comenzar el tratamiento con Xeljanz en pacientes con valores bajos de hemoglobina (es decir, < 9 g/dL). El tratamiento con Xeljanz se debe interrumpir en pacientes que presentan concentraciones de hemoglobina < 8 g/dL o cuya concentración de hemoglobina disminuye > 2 g/dL con el tratamiento. Se debe supervisar la concentración de hemoglobina en el momento basal, después de 4 a 8 semanas de tratamiento y cada 3 meses posteriormente. Lípidos: El tratamiento con Xeljanz se asoció con aumentos en los parámetros lipídicos, por ejemplo, colesterol total, colesterol de lipoproteínas de baja densidad (LDL) y colesterol de lipoproteínas de alta densidad (HDL). Los efectos máximos en general se observaron en el período de 6 semanas. Se deben evaluar los parámetros lipídicos aproximadamente de 4 a 8 semanas después del inicio del tratamiento con Xeljanz. Los pacientes deben tratarse de acuerdo con las pautas clínicas (por ejemplo, programa nacional de educación sobre el colesterol) para el tratamiento de la hiperlipidemia. Los aumentos en el colesterol total y LDL asociados con Xeljanz se reducen a las concentraciones anteriores al tratamiento con la administración de estatinas. Vacunas: No hay datos disponibles sobre la respuesta a las vacunas ni sobre la transmisión secundaria de la infección por vacunas con virus vivos en los pacientes que reciben Xeljanz. Se recomienda no administrar vacunas con virus vivos al mismo tiempo que Xeljanz. Se recomienda que todos los pacientes estén al día con todas las vacunas conforme a las pautas de vacunación actuales antes de comenzar el tratamiento con Xeljanz. Pacientes con insuficiencia renal: No es necesario ajustar la dosis en pacientes con insuficiencia renal leve. La dosis de Xeljanz no debe superar los 5 mg una vez al día en pacientes con insuficiencia renal moderada o grave, Xeljanz no se evaluó en pacientes con valores basales de depuración de creatinina (calculados según la ecuación de Cockroft-Gault) inferiores a 40 mL/min en estudios clínicos. Pacientes con insuficiencia hepática: No es necesario ajustar la dosis en pacientes con insuficiencia hepática leve. La dosis de Xeljanz no debe superar los 5 mg una vez al día en pacientes con insuficiencia hepática moderada. El tratamiento con Xeljanz no está recomendado en pacientes con insuficiencia hepática grave. Xeljanz no se evaluó en pacientes con insuficiencia hepática grave ni en pacientes con serología positiva de HBV y HCV en estudios clínicos. Combinación con DMARD biológicos: El uso de Xeljanz no se ha estudiado y debe evitarse en pacientes con RA en combinación con DMARD biológicos, como antagonistas del TNF, antagonistas de IL-1R, antagonistas de IL-6R, anticuerpos monoclonales anti-CD20 y moduladores selectivos de la coestimulación, y con inmunosupresores potentes, como azatioprina y ciclosporina, debido a la posibilidad de que aumente la inmunosupresión y el riesgo de infecciones. Fertilidad, embarazo y lactancia: No hay estudios adecuados y bien controlados respecto del uso de Xeljanz en mujeres embarazadas. Se ha demostrado que tofacitinib es teratógeno en ratas y conejos, y que tiene efectos en la fertilidad de las hembras, el parto y el desarrollo peri/posnatal en las ratas. Xeljanz no debe usarse durante el embarazo a menos que sea estrictamente necesario. Se encontró tofacitinib en la leche de ratas lactantes. No se sabe si tofacitinib se segrega en la leche materna. Las mujeres no deben amamantar mientras reciben tratamiento con Xeljanz.
Interacciones.
Interacciones que afectan el uso de Xeljanz: Debido a que tofacitinib es metabolizado por el CYP3A4, es probable la interacción con fármacos que inhiben o inducen el CYP3A4. La exposición a tofacitinib aumenta cuando se coadministra con inhibidores potentes del CYP3A4 (por ejemplo, ketoconazol) o cuando la administración de uno o más medicamentos concomitantes causa la inhibición moderada del CYP3A4 y la inhibición potente del CYP2C19 (por ejemplo, fluconazol). La exposición a tofacitinib disminuye cuando se coadministra con inductores potentes del CYP3A4 (por ejemplo, rifampicina). Es improbable que los inhibidores del CYP2C19 o de la glucoproteína P modifiquen la farmacocinética (PK) de tofacitinib. La administración concomitante con metotrexato (de 15 a 25 mg de MTX una vez a la semana) no tuvo efectos en la PK de tofacitinib. La coadministración de ketoconazol, un inhibidor potente del CYP3A4, con una dosis única de tofacitinib aumentó los valores de AUC y la Cmáx en 103% y 16%, respectivamente. La coadministración de fluconazol, un inhibidor moderado del CYP3A4 y un inhibidor fuerte del CYP2C19, aumentó los valores de AUC y la Cmáx de tofacitinib en 79% y 27%, respectivamente. La coadministración de tacrolimus (Tac), un inhibidor leve del CYP3A4, aumentó los valores de AUC de tofacitinib en 21% y disminuyó la Cmáx de tofacitinib en 9%. La coadministración de ciclosporina (CsA), un inhibidor moderado del CYP3A4, aumentó los valores de AUC de tofacitinib en 73% y disminuyó la Cmáx de tofacitinib en 17%. No se ha estudiado el uso combinado de dosis múltiples de tofacitinib con estos potentes inmunosupresores en pacientes con artritis reumatoide. La coadministración de rifampicina, un inductor fuerte del CYP3A4, disminuyó los valores de AUC y la Cmáx de tofacitinib en 84% y 74%, respectivamente. Posibilidad de que Xeljanz influya en la PK de otros fármacos Estudios in vitro indican que tofacitinib no inhibe ni induce de forma significativa la actividad de los principales CYP (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 y CYP3A4) metabolizadores de fármacos en seres humanos en concentraciones que superan 150 veces la Cmáx en estado de equilibrio de una dosis de 10 mg BID. Estos resultados in vitro fueron confirmados por un estudio de interacción farmacológica en seres humanos que no mostró cambios en la PK de midazolam, un sustrato del CYP3A4 muy sensible, cuando se coadministró con tofacitinib. Los datos in vitro indican que la posibilidad de que tofacitinib inhiba a transportadores como la glucoproteína P, transportadores catiónicos orgánicos o aniónicos orgánicos en concentraciones terapéuticas también es baja. La coadministración de tofacitinib no tuvo un efecto en la PK de los anticonceptivos orales, levonorgestrel y etinilestradiol, en voluntarias sanas. La coadministración de tofacitinib con metotrexato, de 15 a 25 mg, una vez a la semana, disminuyó los valores de AUC y la Cmáx de metotrexato en 10% y 13%, respectivamente. El grado de disminución de la exposición al metotrexato no justifica modificaciones en la administración de dosis individuales de metotrexato. En pacientes con artritis reumatoide, la depuración oral de tofacitinib no varía con el tiempo, lo que indica que tofacitinib no normaliza la actividad de la enzima CYP en pacientes con AR. Por lo tanto, no se espera que la coadministración con tofacitinib genere aumentos clínicamente relevantes en el metabolismo de los sustratos del CYP en pacientes con AR. Población pediátrica Se han hecho estudios solamente en adultos.
Sobredosificación.
No hay experiencia de sobredosis de Xeljanz. No hay un antídoto específico para la sobredosis de Xeljanz. El tratamiento debe ser sintomático y de apoyo. En caso de sobredosis, se recomienda supervisar al paciente para detectar signos y síntomas de reacciones adversas. Los pacientes que tienen reacciones adversas deben recibir el tratamiento adecuado. Los datos farmacocinéticos de una dosis única de hasta 100 mg inclusive en voluntarios sanos indican que se espera que más del 95 % de la dosis administrada se elimine en el período de 24 horas.
Presentación.
XELJANZ 5mg x 56 comprimidos recubiertos.