XELODA®
ROCHE
Agente citostático.
Composición.
Principio activo: capecitabina.
Farmacología.
Propiedades farmacodinámicas: Mecanismo de acción: La capecitabina es un carbamato fluoropirimidínico, diseñado como agente citotóxico oral, activado en los tumores y con selectividad para éstos. In vitro, la capecitabina no es citotóxica. In vivo, en cambio, se convierte de manera secuencial en la porción citotóxica 5-fluorouracilo (5-FU), que es a su vez metabolizada. La formación del 5-FU tiene lugar sobre todo en el propio tumor, por acción de la enzima timidina-fosforilasa (dThdPasa), factor angiógeno tumoral; de este modo, la exposición de los tejidos sanos a la acción sistémica del 5-FU queda reducida al mínimo. La biotransformación enzimática secuencial de la capecitabina en 5-FU tiene como consecuencia concentraciones más altas dentro de las células tumorales. Tras la administración oral de la capecitabina a pacientes con carcinoma colorrectal (n = 8), la razón de la concentración de 5-FU en el tumor colorrectal y los tejidos adyacentes era de 3,2 (de 0,9 a 8,0). La razón de la concentración de 5-FU en el tumor y el plasma era de 21,4 (de 3,9 a 59,9), mientras que en los tejidos sanos y el plasma era de 8,9 (de 3,0 a 25,8). La actividad de la timidina-fosforilasa fue 4 veces mayor en el tumor colorrectal primario que en el tejido sano adyacente. Diversos tumores humanos -como el carcinoma de mama, el carcinoma gástrico, el carcinoma colorrectal, el carcinoma cervicouterino y el carcinoma de ovario- presentan valores mayores de timidina-fosforilasa (capaz de transformar la 5'-desoxi-5- fluorouridina [5'-DFUR] en 5-FU) que los tejidos sanos correspondientes. Tanto las células sanas como las células cancerosas metabolizan el 5-FU a monofosfato de 5-fluoro-2-desoxiuridina (FdUMP) y trifosfato de 5-fluorouridina (FUTP). Estos metabolitos provocan daños celulares por dos mecanismos distintos. En primer lugar, el FdUMP y el cofactor N5-10-metilenotetrahidrofolato se unen a la enzima timidilato-sintasa (TS) para formar un complejo ternario con enlace covalente. Esta unión inhibe la formación de timidilato a partir del uracilo. El timidilato es el precursor necesario del trifosfato de timidina, esencial para la síntesis de ADN, de tal modo que la deficiencia de este compuesto puede inhibir la división celular. En segundo lugar, las enzimas transcriptivas nucleares pueden incorporar por error FUTP en lugar de trifosfato de uridina (UTP) durante la síntesis de ARN, y este error metabólico puede interferir con los procesos de interpretación del ARN y síntesis de proteínas. Ensayos clínicos / Eficacia: Carcinoma de colon, carcinoma colorrectal: Monoterapia en el tratamiento adyuvante del carcinoma de colon: Los datos de un ensayo clínico de fase III, multicéntrico, aleatorizado y controlado (estudio XACT: M66001) en pacientes con cáncer de colon en estadio III (estadio C de Dukes) respaldan el empleo de Xeloda para el tratamiento adyuvante de pacientes con cáncer de colon. En este estudio se aleatorizaron 1.987 pacientes para recibir Xeloda (1.250 mg/m2 dos veces al día durante 2 semanas, seguido de una 1 semana de reposo farmacológico, administrado en ciclos de 3 semanas, durante 24 semanas) o 5-FU y leucovorina (régimen de la Clínica Mayo: 20 mg/m2 de leucovorina i.v., seguido de 425 mg/m2 de 5-FU en bolo i.v., los días 1 a 5 de cada 28 días, durante 24 semanas). Xeloda fue al menos equivalente a 5-FU/LV i.v. en cuanto a supervivencia sin enfermedad (p = 0,0001, margen de no inferioridad de 1,2). En toda la población aleatorizada, las pruebas para diferenciar la supervivencia sin enfermedad y la supervivencia global con Xeloda frente a 5-FU/LV arrojaron una hazard ratio (HR, razón de riesgos instantáneos) de 0,88 (IC del 95%: 0,77-1,01; p = 0,068) y de 0,86 (IC del 95%: 0,74-1,01; p = 0,060), respectivamente. La mediana de seguimiento en el momento del análisis era de 6,9 años. Politerapia en el tratamiento adyuvante del carcinoma de colon: Los datos de un ensayo clínico de fase III multicéntrico, aleatorizado y controlado en pacientes con cáncer de colon en estadio III (estadio C de Dukes) respaldan el empleo de Xeloda en asociación con oxaliplatino (XELOX) para el tratamiento adyuvante de los pacientes con cáncer de colon (estudio NO16968). En este estudio se aleatorizó a 944 pacientes para recibir en ciclos de 3 semanas, durante 24 semanas, Xeloda (1.000 mg/m2 dos veces al día durante 2 semanas, seguido de 1 semana de reposo farmacológico) en asociación con oxaliplatino (130 mg/m2 en infusión i.v. de 2 horas en el día 1 de cada 3 semanas); 942 pacientes fueron aleatorizados para recibir 5-FU en bolo i.v. y leucovorina. En el análisis principal de la supervivencia sin enfermedad (SSE) de la población con intención de tratar se puso de manifiesto que XELOX era significativamente superior a 5-FU/LV (HR = 0,80, IC del 95%: 0,69-0,93; p = 0,0045). La tasa de SSE a los 3 años era del 71% con XELOX y del 67% con 5-FU/LV. El análisis de la variable secundaria de valoración, la supervivencia sin recaída, respalda estos resultados con una HR de 0,78 (IC del 95%: 0,67-0,92; p = 0,0024) para XELOX en comparación con 5-FU/LV. XELOX mostró una tendencia hacia una supervivencia global (SG) superior con una HR de 0,87 (IC del 95%: 0,72-1,05; p = 0,1486), que significa una reducción del riesgo de muerte del 13%. La tasa de SG a los 5 años era del 78% con XELOX y del 74% con 5-FU/LV. Los datos de la eficacia proporcionados se basan en una mediana de observación de 59 meses para la SG y de 57 meses para la SSE. Monoterapia en el carcinoma colorrectal metastásico: Los datos de dos ensayos clínicos de fase III de diseño idéntico, controlados, multicéntricos y aleatorizados respaldan el uso de Xeloda como tratamiento de primera línea del carcinoma colorrectal metastásico (SO14695; SO14796). En estos estudios se asignaron aleatorizadamente 603 pacientes al tratamiento con Xeloda (1.250 mg/m2 dos veces al día durante 2 semanas, seguido de una 1 semana de reposo farmacológico, administrado en ciclos de 3 semanas) y 604 pacientes al tratamiento con 5-FU y leucovorina (régimen de la Clínica Mayo: 20 mg/m2 de leucovorina i.v., seguido de 425 mg/m2 de 5-FU en bolo i.v., los días 1 a 5 de cada 28 días). Las tasas de respuesta global objetiva en toda la población aleatorizada (evaluación del investigador) fueron del 25,7% (Xeloda) frente al 16,7% (régimen de la Clínica Mayo); p < 0,0002. La mediana del tiempo hasta la progresión fue de 140 días (Xeloda) frente a 144 días (régimen de la Clínica Mayo). La mediana de la supervivencia fue de 392 días (Xeloda) frente a 391 días (régimen de la Clínica Mayo). Politerapia en el tratamiento de primera línea del carcinoma colorrectal: Los datos de un estudio clínico de fase III, multicéntrico, aleatorizado y controlado (N0 16966) respaldan el uso de Xeloda en asociación con oxaliplatino o en asociación con oxaliplatino y bevacizumab (BV) como tratamiento de primera línea del carcinoma colorrectal metastásico. Este estudio constaba de dos partes: una parte inicial de 2 grupos en la que se distribuyó aleatorizadamente a los pacientes entre dos grupos de tratamiento diferentes, XELOX y FOLFOX-4, y una parte siguiente bifactorial (2x2) con cuatro grupos de tratamiento diferentes: XELOX + placebo (P), FOLFOX-4 + P, XELOX + BV y FOLFOX-4 + BV. En la tabla siguiente se resumen los regímenes terapéuticos.
La no inferioridad de XELOX respecto de FOLFOX-4 en la comparación global de los grupos quedó demostrada en cuanto a la supervivencia sin progresión (SSP) en la población elegible y la población con intención de tratar (ver tabla siguiente). Los resultados indican que XELOX es equivalente a FOLFOX-4 en cuanto a supervivencia global. La comparación de XELOX + bevacizumab con FOLFOX-4 + bevacizumab era un análisis exploratorio preespecificado. En esta comparación de subgrupos de tratamiento, XELOX + bevacizumab fue similar a FOLFOX-4 + bevacizumab en cuanto a supervivencia sin progresión (hazard ratio de 1,01 [IC del 97,5%: 0,84-1,22]). La mediana de seguimiento en el momento de los análisis principales de la población con intención de tratar era de 1,5 años; la tabla siguiente contiene también datos de los análisis después de 1 año más de seguimiento.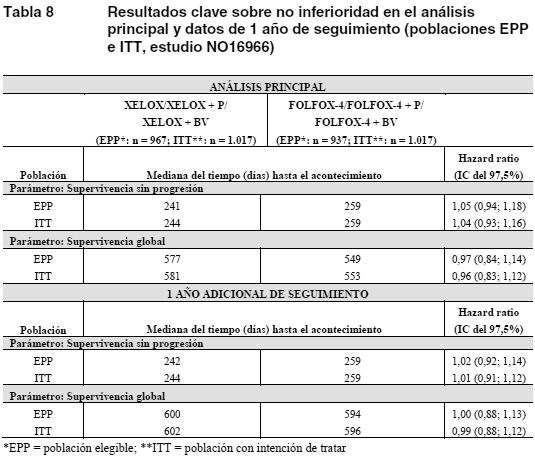
Politerapia en el carcinoma esofagogástrico: Los datos de un ensayo clínico fase III, multicéntrico, aleatorizado y controlado en pacientes con cáncer esofagogástrico avanzado o metastásico respaldan el uso de Xeloda como tratamiento de primera línea del cáncer esofagogástrico avanzado. En este estudio se aleatorizaron 160 pacientes para recibir Xeloda (1.000 mg/m2 dos veces al día, durante 2 semanas, seguido de 7 días de reposo farmacológico) y cisplatino (80 mg/m2 en infusión i.v. de 2 horas, cada 3 semanas). Un total de 156 pacientes fueron aleatorizados al tratamiento con 5-FU (800 mg/m2 al día en infusión continua, los días 1 a 5 de cada 3 semanas) y cisplatino (80 mg/m2 en infusión de 2 horas, el día 1 de cada 3 semanas). El objetivo principal del estudio se alcanzó: en el análisis según protocolo, Xeloda en asociación con cisplatino fue al menos equivalente a la asociación de 5-FU y cisplatino en cuanto a supervivencia sin progresión. El resultado en cuanto a duración de la supervivencia (supervivencia global) fue similar al de supervivencia sin progresión de la enfermedad (ver tabla siguiente).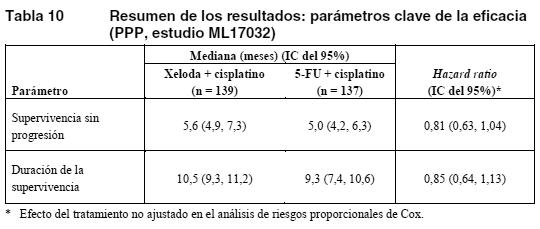
Los datos de un ensayo clínico fase III aleatorizado y multicéntrico para comparar capecitabina con 5-FU y oxaliplatino con cisplatino en pacientes con carcinoma gástrico avanzado respaldan el uso de Xeloda como tratamiento de primera línea del carcinoma gástrico avanzado. En este estudio se aleatorizó a 1.002 pacientes según un diseño bifactorial (2x2) entre los 4 grupos siguientes: ECF: epirrubicina (50 mg/m2 en bolo i.v. el día 1 de cada 3 semanas), cisplatino (60 mg/m2 en infusión de dos horas el día 1 de cada 3 semanas) y 5-FU (200 mg/m2 al día en infusión continua por una vía central). ECX: epirubicina (50 mg/m2 en bolo i.v. el día 1 de cada 3 semanas), cisplatino (60 mg/m2 en infusión de dos horas el día 1 de cada 3 semanas) y Xeloda (625 mg/m2 dos veces al día continuamente). EOF: epirubicina (50 mg/m2 en bolo i.v. el día 1 de cada 3 semanas), oxaliplatino (130 mg/m2 en infusión de dos horas el día 1 de cada 3 semanas) y 5-FU (200 mg/m2 al día en infusión continua por una vía central). EOX: epirubicina (50 mg/m2 en bolo i.v. el día 1 de cada 3 semanas), oxaliplatino (130 mg/m2 en infusión de dos horas el día 1 de cada 3 semanas) y Xeloda (625 mg/m2 dos veces al día continuamente). El análisis principal de la eficacia en la población según protocolo demostró la no inferioridad en cuanto a supervivencia global de los regímenes basados en la capecitabina frente a los basados en el 5-FU (hazard ratio de 0,86, IC del 95%: 0,8-0,99) y de los basados en el oxaliplatino frente a los basados en el cisplatino (hazard ratio de 0,92, IC del 95%: 0,8-1,1). La mediana de la supervivencia global fue de 10,9 meses con los regímenes basados en la capecitabina y de 9,6 meses con los basados en el 5-FU. La mediana de la supervivencia global fue de 10,0 meses con los regímenes basados en el cisplatino y de 10,4 meses con los basados en el oxaliplatino. Xeloda también se ha utilizado en asociación con oxaliplatino como tratamiento del carcinoma gástrico avanzado. Los estudios de Xeloda en monoterapia muestran que este producto es activo en el carcinoma gástrico avanzado. Carcinoma de colon, colorrectal y esofagogástrico avanzado: metanálisis: Los resultados de un metanálisis de seis estudios clínicos (SO14695, SO14796, M66001, NO16966, NO16967 y M17032) respaldan la sustitución de 5-FU por Xeloda en el tratamiento monoterápico y politerápico del cáncer gastrointestinal. En el análisis de datos agrupados se incluyó a 3.097 pacientes tratados con los regímenes que contenían Xeloda y a 3.074 pacientes de los regímenes con 5-FU. La hazard ratio de la supervivencia global fue de 0,9694 (IC del 95%: 0,9089-1,0200, p = 0,0489), lo cual muestra que los regímenes con Xeloda son superiores a los que contienen 5-FU. Politerapia en el carcinoma de mama: Los datos de un estudio clínico fase III, controlado, multicéntrico y aleatorizado respaldan el uso de Xeloda en asociación con docetaxel para el tratamiento del carcinoma de mama localmente avanzado o metastático tras el fracaso de una quimioterapia citotóxica que incluya una antraciclina. En ese estudio, se aleatorizaron 255 pacientes para recibir Xeloda (1.250 mg/m2 dos veces al día durante 2 semanas, seguido de 1 semana de reposo farmacológico) y docetaxel (75 mg/m2 en infusión i.v. de 1 hora cada 3 semanas). Un total de 256 pacientes fueron aleatorizados para recibir docetaxel solo (100 mg/m2 en infusión i.v. de 1 hora cada 3 semanas). La supervivencia fue mayor en el grupo de tratamiento con Xeloda + docetaxel (p = 0,0126). La mediana de la supervivencia fue de 442 días (Xeloda + docetaxel) frente a 352 días (docetaxel solo). La tasa global de respuesta objetiva en toda la población aleatorizada (evaluación del investigador) fue del 41,6% (Xeloda + docetaxel) frente al 29,7% (docetaxel solo); p < 0,0058. El tiempo hasta la progresión de la enfermedad o la muerte fue superior en el grupo con Xeloda + docetaxel (p < 0,0001). La mediana del tiempo hasta la progresión fue de 186 días (Xeloda + docetaxel) frente a 128 días (docetaxel solo). Monoterapia en el carcinoma de mama: Los datos de dos estudios de fase II multicéntricos respaldan el uso de Xeloda en monoterapia contra el carcinoma de mama localmente avanzado o metastásico cuando haya fracasado la quimioterapia con taxanos o antraciclinas o cuando no esté indicado proseguir el tratamiento con antraciclinas. En estos estudios, un total de 236 pacientes recibieron tratamiento con Xeloda (1.250 mg/m2 dos veces al día durante 2 semanas, seguido de 1 semana de reposo farmacológico). La tasa global de respuesta objetiva (evaluación del investigador) fue del 20% (primer estudio) y 25% (segundo estudio). La mediana del tiempo hasta la progresión fue de 93 y 98 días. La mediana de supervivencia fue de 384 y 373 días. Propiedades farmacocinéticas: Absorción: Administrada por vía oral, la capecitabina se absorbe de forma rápida y extensa, para experimentar a continuación una amplia conversión a los metabolitos 5'-desoxi-5- fluorocitidina (5'-DFCR) y 5'-DFUR. La administración con alimentos reduce la velocidad de absorción de la capecitabina, pero sólo afecta mínimamente al ABC de la 5'-DFUR y del metabolito posterior 5-FU. Con la dosis de 1.250 mg/m2 el día 14 administrada después de tomar alimentos, las concentraciones plasmáticas máximas (Cmáx en mg/ml) de capecitabina, 5'-DFCR, 5'-DFUR, 5-FU y FBAL fueron de 4,67, 3,05, 12, 1, 0,95 y 5,46, respectivamente. El tiempo hasta la concentración plasmática máxima (Tmáx en horas) fue de 1,50, 2,00, 2,00, 2,00 y 3,34. Los valores de ABC0∞8 en m.h/ml fueron de 7,75, 7,24, 24,6, 2,03 y 36,3. Distribución: Unión a las proteínas: Los estudios in vitro con plasma humano han revelado que la capecitabina, la 5'-DFCR, la 5'-DFUR y el 5-FU se unen en un 54%, 10%, 62% y 10%, respectivamente, a las proteínas plasmáticas, sobre todo a la albúmina. Metabolismo: En una primera etapa, la capecitabina es metabolizada por la carboxilesterasa hepática a 5'-DFCR, la cual se transforma después en 5'-DFUR por efecto de la citidinadesaminasa, localizada fundamentalmente en el hígado y en los tejidos tumorales. La formación de 5-FU tiene lugar sobre todo en el propio tumor, por acción del factor angiógeno tumoral dThdPasa; de este modo, la exposición de los tejidos sanos a la acción sistémica del 5-FU queda reducida al mínimo. El ABC del 5-FU es de 6 a 22 veces menor que el registrado tras su administración en bolo i.v. (dosis de 5-FU de 600 mg/m2). Los metabolitos de la capecitabina únicamente adquieren capacidad citotóxica tras su conversión a 5-FU y los anabolitos de éste (ver Mecanismo de acción). Posteriormente, el 5-FU es catabolizado por la enzima dihidropirimidin-dehidrogenasa (DPD) a los metabolitos inactivos dihidro-5-fluorouracilo (FUH2), ácido 5-fluoroureidopropiónico (FUPA) y a-fluoro-(3-alanina (FBAL). La actividad dihidropirimidindehidrogenasa (DPD) es el paso limitante. Eliminación: La semivida de eliminación (t1/2 en horas) de la capecitabina, 5'-DFCR, 5'-DFUR, 5-FU y FBAL era de 0,85, 1,11, 0,66, 0,76 y 3,23, respectivamente. Se ha estudiado la farmacocinética de la capecitabina en dosis de 502 a 3.514 mg/m2/día. Los parámetros farmacocinéticos de la capecitabina, la 5'-DFCR y la 5'-DFUR eran similares en los días 1 y 14. El ABC de 5-FU era un 30-35% superior en el día 14, pero no aumentó más después (día 22). En dosis terapéuticas, la farmacocinética de la capecitabina y sus metabolitos, a excepción del 5-FU, era proporcional a la dosis. Tras administrar Xeloda por vía oral, los metabolitos de la capecitabina se recuperan principalmente en la orina. La mayor parte (95,5%) de la dosis de capecitabina se recupera en la orina. La excreción fecal es mínima (2,6%). El principal metabolito excretado en la orina es FBAL, que representa un 57% de la dosis administrada. Alrededor del 3% de la dosis administrada se excreta inalterada en la orina. Politerapia: Los estudios fase I para evaluar el impacto de Xeloda en la farmacocinética del docetaxel y el paclitaxel, y viceversa, no mostraron ningún efecto de Xeloda en la farmacocinética del docetaxel y el paclitaxel (Cmáx y ABC) ni del docetaxel y el paclitaxel en la farmacocinética del 5'-DFUR (el principal metabolito de la capecitabina). Farmacocinética en poblaciones especiales: Se ha realizado un análisis de farmacocinética poblacional tras el tratamiento con Xeloda, en una dosis de 1.250 mg/m2 dos veces al día, de 505 pacientes con carcinoma colorrectal. El sexo, la presencia o ausencia de metástasis hepáticas basales, el índice de Karnofsky, la bilirrubina total, la albúmina sérica, ASAT y ALAT no tuvieron ningún efecto estadísticamente significativo en la farmacocinética de 5'-DFUR, 5-FU y FBAL. Pacientes con insuficiencia hepática debida a metástasis hepáticas: En pacientes con cáncer con insuficiencia hepática leve o moderada debida a la presencia de metástasis hepáticas no se ha observado ningún efecto de interés clínico sobre la bioactivación o la farmacocinética de la capecitabina (ver Pautas posológicas especiales). No existen datos farmacocinéticos de pacientes con insuficiencia hepática grave. Pacientes con insuficiencia renal: De acuerdo con un estudio farmacocinético en pacientes con cáncer con insuficiencia renal ligera o grave, no hay indicios de que el aclaramiento de creatinina influya en la farmacocinética de la capecitabina o el 5-FU. El aclaramiento de creatinina sí influyó en la exposición sistémica a la 5'-DFUR (aumento del ABC en un 35% cuando el aclaramiento de creatinina disminuía a la mitad) y la FBAL (aumento del ABC en un 114% cuando el aclaramiento de creatinina disminuía a la mitad). La FBAL es un metabolito sin actividad antiproliferativa; la 5'-DFUR es el precursor directo del 5-FU (ver Pautas posológicas especiales). Ancianos: De acuerdo con un análisis de farmacocinética poblacional en pacientes de muy diversas edades (de 27 a 86 años), incluidos 234 (46%) con más de 65 años, la edad no influye en la farmacocinética de la 5'-DFUR y el 5-FU. El ABC de la FBAL se incrementó con la edad (un aumento de la edad del 20% se traduce en un incremento del ABC de la FBAL del 15%). Este incremento probablemente se debe a cambios en la función renal (ver Pautas posológicas especiales y Farmacocinética en poblaciones especiales, subapartado Pacientes con insuficiencia renal). Raza: En un análisis de farmacocinética poblacional en el que se incluyó a 455 pacientes de raza blanca (90,1%), 22 de raza negra (4,4%) y 28 de otras razas o etnias (5,5%), no se apreciaron diferencias farmacocinéticas entre los pacientes de raza blanca y los de raza negra.
Indicaciones.
Carcinoma de mama: Xeloda en asociación con docetaxel está indicado para el tratamiento del carcinoma de mama localmente avanzado o metastásico tras el fracaso de la quimioterapia citotóxica. El tratamiento anterior debe haber incluido una antraciclina. En monoterapia, Xeloda está indicado para el tratamiento del carcinoma de mama localmente avanzado o metastásico cuando haya fracasado la quimioterapia con taxanos y antraciclinas o cuando no esté indicado proseguir el tratamiento con antraciclinas. Carcinoma colorrectal: Xeloda está indicado como tratamiento adyuvante del cáncer de colon etapa III (etapa C de Dukes) en pacientes quienes han sufrido resección completa del tumor primario. Xeloda está indicado para el tratamiento del cáncer colorrectal metastásico. Carcinoma esofagogástrico: Xeloda está indicado como tratamiento de primera línea del cáncer esofagogástrico avanzado o metastásico, en asociación con epirubicina y oxaliplatino o cisplatino.
Dosificación.
Dosis habitual: Los comprimidos de Xeloda deben ingerirse enteros, con agua, dentro de los 30 minutos siguientes a una comida. Monoterapia: La dosis inicial recomendada de Xeloda en monoterapia es de 1.250 mg/m2 dos veces al día (mañana y noche; equivalente a una dosis diaria total de 2.500 mg/m2) durante 2 semanas, seguido de 7 días de reposo farmacológico. En asociación con docetaxel: En asociación con docetaxel, la dosis recomendada de Xeloda es de 1.250 mg/m2 dos veces al día durante 2 semanas, seguido de 7 días de reposo farmacológico, y la del docetaxel, de 75 mg/m2 en infusión intravenosa (i.v.) de 1 hora, cada 3 semanas. De acuerdo con la información sobre el docetaxel, en los pacientes tratados con Xeloda junto con docetaxel debe iniciarse una premedicación antes de administrarse el docetaxel. En asociación con cisplatino: En asociación con cisplatino, la dosis recomendada de Xeloda es de 1.000 mg/m2 dos veces al día durante 2 semanas, seguido de 7 días de reposo farmacológico. El cisplatino se administra en una dosis de 80 mg/m2 en infusión i.v. de 2 horas el día 1 de cada 3 semanas. La primera dosis de Xeloda se administra por la noche del día 1, y la última dosis, por la mañana del día 15. De acuerdo con la información sobre el cisplatino, en los pacientes tratados con Xeloda junto con cisplatino debe iniciarse una premedicación antiemética y suficientemente hidratante antes de administrarse el cisplatino. En asociación con oxaliplatino y/o bevacizumab: En asociación con oxaliplatino y/o bevacizumab, la dosis recomendada de Xeloda es de 1.000 mg/m2 dos veces al día durante 2 semanas, seguido de 7 días de reposo farmacológico. La primera dosis de Xeloda se administra en la víspera del día 1, y la última dosis, por la mañana del día 15. En una pauta de una vez (el día 1) cada 3 semanas, el bevacizumab se administra en infusión i.v. de 7,5 mg/kg durante 30 a 90 minutos, seguido de oxaliplatino en infusión i.v. de 7,5 mg/kg durante 2 horas. En asociación con oxaliplatino y epirrubicina: La dosis recomendada de Xeloda es de 625 mg/m2 dos veces al día durante 24 semanas sin interrupción, en combinación con 130 mg/m2 (por infusión intravenosa de 2 ó más horas) de oxaliplatino (cada 3 semanas. máximo 8 ciclos) y 50 mg/m2 de epirrubicina por bolo i.v. (cada 3 semanas, máximo 8 ciclos). Para obtener más información de la premedicación para mantener una hidratación adecuada y el tratamiento antiemético antes de la administración de oxaliplatino, referirse a la información disponible para ese producto. En asociación con cisplatino y epirrubicina: La dosis recomendada de Xeloda es de 625 mg/m2 dos veces al día durante 24 semanas sin interrupción, en combinación con 60 mg/m2 de cisplatino (cada 3 semanas, máximo 8 ciclos) y 50 mg/m2 de epirrubicina por bolo i.v. (cada 3 semanas, máximo 8 ciclos). De acuerdo con la información sobre cisplatino, en los pacientes tratados con Xeloda junto con cisplatino debe iniciarse premedicación antiemética y suficientemente hidratante antes de administrarse cisplatino.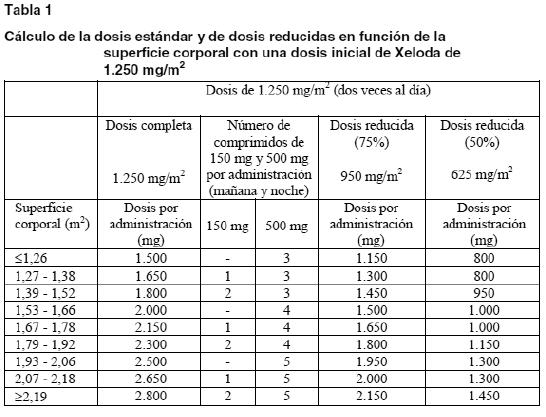
Ajustes posológicos durante el tratamiento: Información general: La toxicidad de Xeloda se puede controlar mediante tratamiento sintomático y/o modificando la dosis de Xeloda (interrupción de la medicación o reducción posológica). Una vez reducida la dosis, no deberá incrementarse después en ningún momento. Si el médico considera improbable que un efecto tóxico pueda volverse grave o poner en peligro la vida del paciente, podrá continuar el tratamiento con la misma dosis sin reducción o interrupción. En presencia de acontecimientos adversos de grado 1 no se recomienda ajustar la dosis. Si los acontecimientos adversos son de grados 2 o 3, debe interrumpirse la administración de Xeloda. Una vez que el acontecimiento adverso haya desaparecido o disminuido de intensidad al grado 1, puede restablecerse el tratamiento con la dosis completa de Xeloda o, si es preciso, ajustada según la Tabla 3. De presentarse una reacción de grado 4, se retirará Xeloda hasta su resolución o disminución al grado 1, para reinstaurar después su administración con la mitad de la dosis original. A los pacientes en tratamiento con Xeloda se les debe informar sobre la necesidad de interrumpir inmediatamente la administración si experimentan toxicidad moderada o grave. No se sustituirán las dosis de Xeloda omitidas por toxicidad. Hematología: No se administrará Xeloda a pacientes con un recuento basal de neutrófilos < 1,5 x 109/l y/o de plaquetas < 100 x 109/l. Si un análisis de laboratorio no programado durante un ciclo de tratamiento revela toxicidad hematológica de grado 3 o 4, se retirará Xeloda. La tabla siguiente recoge las modificaciones posológicas recomendadas en caso de toxicidad asociada a Xeloda.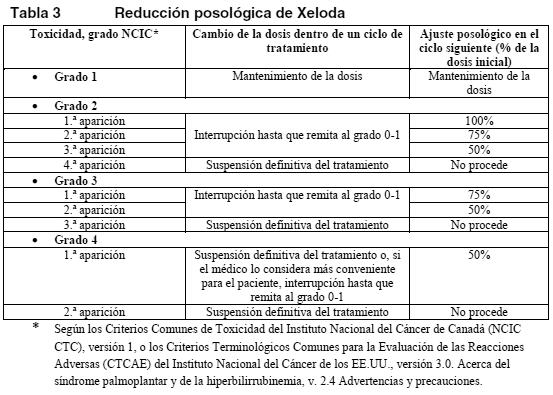
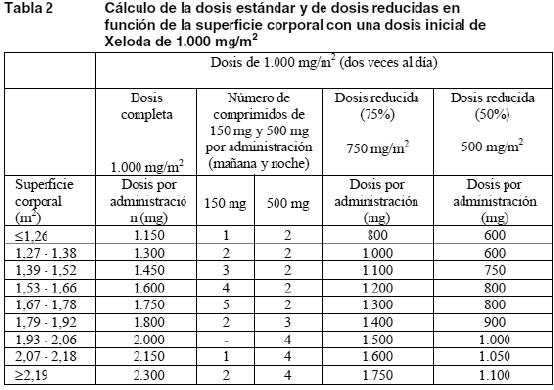
Politerapia general: Los cambios posológicos por toxicidad cuando Xeloda se utilice en asociación con otros tratamientos deben realizarse según la Tabla 3 por lo que respecta a Xeloda y de acuerdo con la información para el prescriptor del otro u otros compuestos. Si al comienzo de un ciclo de tratamiento debe posponerse la administración de Xeloda o del otro u otros compuestos, se pospondrá la administración de todos los medicamentos hasta que se den los requisitos para comenzar con todos. Si el médico considera que la toxicidad durante un ciclo de tratamiento no está relacionada con Xeloda, debe mantenerse la administración de Xeloda y ajustarse la dosis del otro u otros medicamentos de acuerdo con la respectiva información para el prescriptor. Si es necesario retirar definitivamente el otro u otros compuestos, se podrá reanudar el tratamiento con Xeloda cuando se den las condiciones para ello. Esta recomendación es válida para todas las indicaciones y todas las poblaciones especiales. Pautas posológicas especiales: Pacientes con insuficiencia hepática debida a metástasis hepáticas: En caso de insuficiencia hepática leve o moderada a causa de metástasis hepáticas, no es necesario ajustar la dosis inicial. Ahora bien, a tales pacientes se los debe vigilar estrechamente (ver Farmacocinética en poblaciones especiales y Advertencias). En pacientes con insuficiencia hepática grave no se han realizado estudios. Insuficiencia renal: En los pacientes con una insuficiencia renal basal moderada (aclaramiento de creatinina de 30-50 ml/min [Cockroft y Gault]) se recomienda disminuir la dosis de Xeloda al 75% de la dosis inicial de 1.250 mg/m2. En los pacientes con un ligero deterioro de la función renal (aclaramiento de creatinina de 51-80 ml/min) no se recomienda reducir la dosis inicial. Si se presenta algún acontecimiento adverso de grado 2, 3 o 4, se recomienda interrumpir inmediatamente el tratamiento y mantener en estrecha vigilancia al paciente; después, se ajustará la dosis como se indica en la tabla 3 (ver Farmacocinética en poblaciones especiales). Si el aclaramiento calculado de creatinina desciende durante el tratamiento por debajo de 30 ml/min, se retirará Xeloda. Las recomendaciones sobre el ajuste de la dosis en pacientes con deterioro moderado de la función renal son válidas tanto para la monoterapia como para el uso politerápico. Para el cálculo de la dosis, ver Tablas 1 y 2. Niños: No se han estudiado la seguridad y la eficacia de Xeloda en los niños. Ancianos: No se necesita ajustar la dosis inicial de Xeloda en monoterapia. Ahora bien, los efectos adversos de grados 3 o 4 relacionados con el tratamiento han sido más frecuentes en pacientes de más de 80 años que en los más jóvenes. Cuando se ha utilizado Xeloda en asociación con otros compuestos, los pacientes ancianos (≥ 65 años) han experimentado más reacciones adversas (RA) de grados 3 o 4 y más RA que han obligado a suspender la administración que los pacientes más jóvenes. Por tanto, se aconseja una cuidadosa vigilancia de los pacientes ancianos. En asociación con docetaxel: se ha observado una mayor incidencia de acontecimientos adversos de grados 3 o 4 relacionados con el tratamiento y de acontecimientos adversos graves relacionados con el tratamiento entre los pacientes de 60 o más años. Para los pacientes de 60 o más años tratados con la asociación de Xeloda y docetaxel se aconseja empezar el tratamiento con una dosis de Xeloda reducida al 75% (950 mg/m2 dos veces al día). Para el cálculo de la dosis, ver Tabla 2. Xeloda en asociación con irinotecán: en los pacientes de 65 o más años se recomienda reducir la dosis inicial de Xeloda a 800 mg/m2 dos veces al día.
Contraindicaciones.
Xeloda está contraindicado en pacientes alérgicos a la capecitabina o a cualquier otro de sus componentes. Xeloda está contraindicado en pacientes con antecedentes de reacciones graves o imprevistas a las fluoropirimidinas o alérgicas al fluorouracilo. Al igual que otras fluoropirimidinas, Xeloda está contraindicado en pacientes con deficiencia probada de dihidropirimidina deshidrogenasa (DPD). Xeloda no debe administrarse simultáneamente con sorivudina o sus análogos químicamente afines, como la brivudina (ver Interacciones). Xeloda está contraindicado en los pacientes con insuficiencia renal grave (aclaramiento de creatinina < 30 ml/min). Si existen contraindicaciones de alguno de los compuestos de un régimen politerápico, ese compuesto no debe utilizarse.
Reacciones adversas.
Ensayos clínicos: Las RA consideradas por el investigador como posible, probable o remotamente relacionadas con Xeloda corresponden a estudios clínicos realizados con Xeloda en monoterapia (tratamiento adyuvante en el carcinoma de colon, el carcinoma colorrectal metastásico y el carcinoma de mama metastásico) y estudios clínicos con Xeloda en asociación con diversos regímenes quimioterápicos para múltiples indicaciones. Las RA se han categorizado en las tablas siguientes de acuerdo con la incidencia más alta hallada en el análisis de los datos agrupados de siete estudios clínicos. Las RA están enumeradas dentro de cada grupo de frecuencia por orden descendente de gravedad. Las frecuencias se definen como muy frecuentes ≥ 1/10, frecuentes ≥ 5/100 a < 1/10 o infrecuentes ≥ 1/1.000 a < 1/100. Xeloda en monoterapia: Se notificaron los datos de seguridad de Xeloda en monoterapia de los pacientes que habían recibido tratamiento adyuvante del cáncer de colon y de los tratados de carcinoma de mama metastásico o carcinoma colorrectal metastásico. La información relativa a la seguridad comprende datos de un estudio de fase III en el tratamiento adyuvante del carcinoma de colon (995 pacientes tratados con Xeloda y 974 tratados con 5-FU/LV i.v.), de 4 estudios de fase II en mujeres con carcinoma de mama (n = 319) y de 3 estudios (1 de fase II y 2 de fase III) en hombres y mujeres con carcinoma colorrectal (n = 630). El perfil de seguridad de Xeloda en monoterapia era comparable en los pacientes que habían recibido tratamiento adyuvante del carcinoma de colon y en los tratados de carcinoma de mama metastásico o carcinoma colorrectal metastásico. La intensidad de las RA se clasificó según las categorías de toxicidad del sistema NCIC-CTC.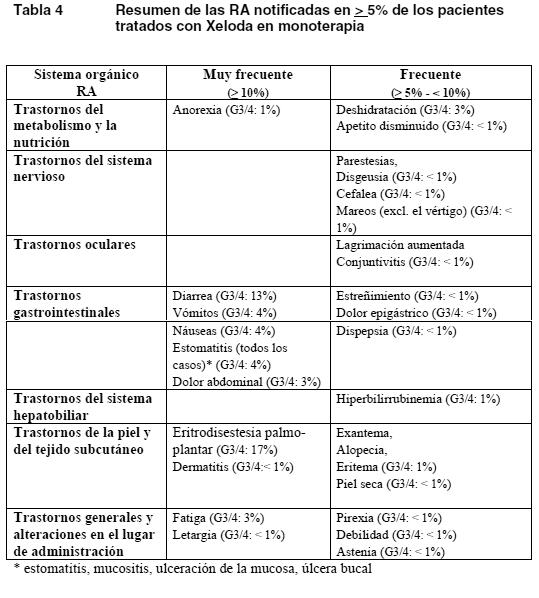
Fisuras cutáneas como mínimo remotamente relacionadas con Xeloda se notificaron en menos del 2% de los pacientes de siete estudios clínicos terminados (n = 949). La siguiente lista de RA, todas ellas bien conocidas con los tratamientos fluoropirimidínicos, se notificaron como al menos remotamente relacionadas con Xeloda en menos del 5% de los 949 pacientes incluidos en siete estudios clínicos ya terminados: Trastornos gastrointestinales: boca seca, flatulencia, RA relacionadas con la inflamación o la ulceración de las mucosas (como esofagitis, gastritis, duodenitis, colitis y hemorragia digestiva). Trastornos cardíacos: edema de las extremidades inferiores, dolor precordial (incluida la angina de pecho), miocardiopatía, isquemia miocárdica (o infarto agudo de miocardio), insuficiencia cardíaca, muerte súbita, taquicardia, arritmias auriculares (incluida la fibrilación auricular) y extrasistolia ventricular. Trastornos neurológicos: disgeusia, insomnio, confusión, encefalopatía, signos cerebelosos (por ejemplo; ataxia, disartria, trastorno del equilibrio, descoordinación). Infecciones e infestaciones: RA relacionadas con mielodepresión, inmunodepresión o alteración de las barreras mucosas, como infecciones locales y generales fatales (de origen bacteriano, vírico o fúngico) y sepsis. Trastornos de la sangre y del sistema linfático: anemia, depresión de la médula ósea, pancitopenia. Trastornos de la piel y del tejido subcutáneo: prurito, exfoliación localizada, hiperpigmentación cutánea, trastornos de las uñas, reacciones de fotosensibilidad, síndrome de posradiación. Trastornos generales y reacciones en el sitio de administración: astenia, dolor en las extremidades, letargia, dolor torácico (no cardíaco). Ojos: irritación ocular. Aparato respiratorio: disnea, tos. Aparato locomotor: dolor de espalda, mialgia, artralgia. Trastornos psíquicos: depresión. Se han descrito casos de insuficiencia hepática y hepatitis colestásica tanto en la fase de investigación clínica como después de la comercialización, pero sin que se estableciera una relación causal con el tratamiento con Xeloda. Xeloda en politerapia: La Tabla 5 recoge las RA asociadas a Xeloda en combinación con diversos regímenes quimioterápicos en múltiples indicaciones y que se produjeron además de las observadas con el uso en monoterapia y/o con una frecuencia mayor. El perfil de seguridad era similar en todas las indicaciones y con todas las pautas politerápicas. Estas reacciones se produjeron en al menos el 5% de los pacientes tratados con Xeloda en combinación con otros quimioterápicos. Las reacciones adversas se han categorizado en esta tabla de acuerdo con la incidencia más alta registrada en cualquiera de los estudios clínicos principales. Algunas de las reacciones adversas son habituales en la quimioterapia (por ejemplo: neuropatía sensorial periférica con docetaxel u oxaliplatino, hipertensión observada con bevacizumab); ahora bien, no cabe excluir la agudización en el tratamiento con Xeloda.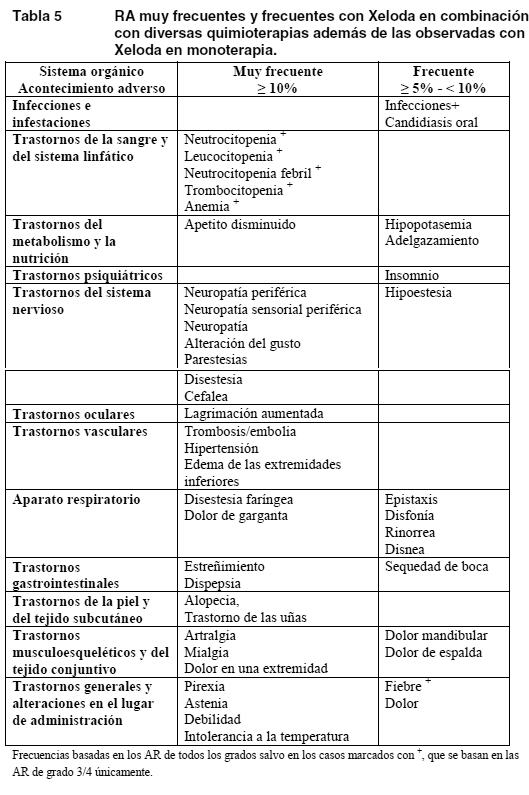
Se han descrito con frecuencia reacciones de hipersensibilidad (2%) e isquemia cardíaca/infarto cardíaco (3%) con Xeloda en combinación con otros quimioterápicos, pero en menos del 5% de los pacientes. Las RA con una frecuencia escasa o infrecuentes notificadas con Xeloda en combinación con otros quimioterápicos concuerdan con las notificadas con Xeloda en monoterapia o el producto usado en combinación en monoterapia (ver información para el prescriptor del producto usado en combinación). Alteraciones analíticas: En la tabla siguiente se recogen las alteraciones analíticas observadas en 995 pacientes (tratamiento adyuvante de carcinoma de colon) y 949 (carcinoma metastásico de mama o colorrectal), independientemente de su relación con la administración de Xeloda.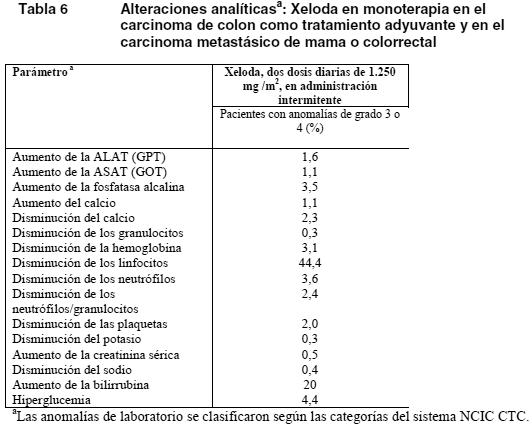
Experiencia tras la comercialización: Las RA indicadas a continuación se han notificado tras la comercialización: Frecuencia muy escasa: estenosis del conducto lacrimal, sin especificar. Frecuencia muy escasa: Se han descrito casos de insuficiencia hepática y hepatitis colestásica durante la investigación clínica y después de la comercialización.
Advertencias.
Advertencias: Diarrea: Xeloda puede provocar diarrea, en ocasiones intensa. Se debe vigilar cuidadosamente a los pacientes con diarrea intensa y, si llegaran a deshidratarse, se les administrarán líquidos y electrólitos. Se debe iniciar lo antes posible un tratamiento antidiarreico estándar (por ejemplo: con loperamida) médicamente adecuado. Si es preciso, debe reducirse la dosis (ver Dosificación). Deshidratación: La deshidratación se debe prevenir o corregir desde el principio. Los pacientes con anorexia, astenia, náuseas, vómitos o diarrea pueden deshidratarse más rápidamente. Si se observa deshidratación de grado 2 (o mayor), se interrumpirá inmediatamente el tratamiento con Xeloda y se tomarán medidas para corregir la deshidratación. No se reanudará el tratamiento hasta que no esté rehidratado el paciente y se hayan corregido o controlado las causas desencadenantes. Si es necesario, se ajustará la dosis en función de la causa desencadenante (ver Dosificación). Precauciones: El patrón de cardiotoxicidad observado con Xeloda es semejante al descrito para otras pirimidinas fluoradas: infarto agudo de miocardio, angina de pecho, arritmia cardíaca, parada cardíaca, insuficiencia cardíaca y alteraciones electrocardiográficas. Estas reacciones adversas pueden ser más frecuentes en los pacientes con antecedentes de coronariopatía. En raras ocasiones se han descrito efectos secundarios graves e imprevistos asociados al 5-FU (por ejemplo: estomatitis, diarrea, neutrocitopenia y neurotoxicidad) y atribuidos a una deficiencia de dihidropirimidina-deshidrogenasa (DPD). Así pues, no cabe excluir una relación entre la deficiencia de DPD y un aumento de los efectos tóxicos potencialmente mortales del 5-FU. Xeloda puede inducir el síndrome palmoplantar (también conocido como síndrome mano-pie, eritrodisestesia palmoplantar o eritema acral inducido por quimioterapia), un efecto tóxico cutáneo. En los pacientes tratados con Xeloda en monoterapia por enfermedad metastásica, la median