YERVOY*
BMS
Tratamiento de melanoma metastásico o no extirpable. Antineoplásico.
Descripción.
YERVOY (ipilimumab) es un anticuerpo monoclonal humano recombinante que se une al antígeno 4 asociado a linfocitos T citotóxicos (CTLA-4). El ipilimumab es una inmunoglobulina IgG1 K con un peso molecular aproximado de 148 kDa. El ipilimumab se produce en los cultivos celulares de los mamíferos (ovario de hámster chino). YERVOY es una solución estéril, sin preservantes, transparente a ligeramente opalescente, incolora a amarilla pálida para infusión intravenosa, que puede contener una pequeña cantidad de partículas de ipilimumab amorfas visiblemente translúcidas a blancas.
Composición.
YERVOY (ipilimumab) se suministra en frascos ampolla/viales de un solo uso de 50 mg/10 mL y 200 mg/40 mL. Cada frasco ampolla/vial de 10 mL contiene 50 mg de ipilimumab. Cada frasco ampolla/vial de 40 mL contiene 200 mg de ipilimumab. Cada mililitro contiene 5 mg de ipilimumab y los siguientes excipientes: tris clorhidrato, cloruro de sodio, manitol, ácido dietilentriaminopentaacético (DTPA), polisorbato 80 (de origen vegetal) y agua para inyección, USP con pH 7.0.
Farmacología.
Mecanismo de acción: La CTLA-4 es un regulador negativo de la activación de las células T. El ipilimumab se fija a la CTLA-4 y bloquea la interacción de la CTLA-4 con sus ligandos, CD80/CD86. Se ha demostrado que el bloqueo de la CTLA-4 incrementa la activación y proliferación de las células T. El mecanismo de acción del efecto del ipilimumab en los pacientes con melanoma es indirecto, posiblemente a través de las respuestas inmunitarias antitumorales mediadas por las células T. Farmacocinética: La farmacocinética del ipilimumab se estudió en 785 pacientes con melanoma no extirpable o metastásico que recibieron dosis de 0,3; 3 o 10 mg/kg una vez cada 3 semanas con un total de cuatro dosis. La concentración máxima (Cmáx), la concentración mínima (Cmín), y el área bajo la curva de concentración plasmática en función del tiempo (AUC) del ipilimumab aumentaron de manera proporcional a la dosis dentro del rango de dosis examinado. Luego de dosis repetidas cada 3 semanas, se determinó que el clearance (CL) del ipilimumab era temporalmente constante, y la acumulación sistémica era de 1,5 veces o menos. Las concentraciones en estado estacionario de ipilimumab se alcanzaron a la tercera dosis; la Cmín media en estado estacionario fue de 19,4 mcg/mL luego de dosis repetidas de 3 mg/kg. El valor medio (coeficiente de variación %) generado a través de un análisis farmacocinético poblacional para la vida media terminal t1/2 fue de 15,4 días (34%) y para el clearance fue de 16,8 mL/h (38)%. Poblaciones específicas: Los efectos de varias covariables de la farmacocinética de ipilimumab se evaluaron en análisis farmacocinéticos de la población. El clearance del ipilimumab aumentó junto con el incremento del peso corporal; no obstante, no se recomienda un ajuste de la dosis para el peso corporal luego de la administración sobre una base de mg/kg. Los siguientes factores no tuvieron efectos clínicamente importantes sobre el clearance de ipilimumab: edad (rango de 23 a 88 años), sexo, estado general, insuficiencia renal, insuficiencia hepática leve, terapia antineoplásica previa y niveles basales de lactato deshidrogenasa (LDH). El efecto de la raza no se examinó debido a una cantidad limitada de datos disponibles en grupos étnicos no caucásicos. Insuficiencia renal: El efecto de la insuficiencia renal sobre el clearance del ipilimumab se evaluó en pacientes con insuficiencia renal leve (GFR < 90 y ≥60 mL/min/1,73 m2; n=349), moderada (GFR < 60 y ≥30 mL/min/1,73 m2; n=82) o severa (GFR < 30 y ≥15 mL/min/1,73 m2; n=4) en comparación con pacientes con función renal normal (GFR ≥90 mL/min/1,73 m2; n=350) en análisis farmacocinéticos poblacionales. No se hallaron diferencias clínicamente importantes en el clearance del ipilimumab entre pacientes con insuficiencia renal y pacientes con una función renal normal (ver Uso en Poblaciones Específicas). Insuficiencia hepática: El efecto de la insuficiencia hepática sobre el clearance del ipilimumab se evaluó en pacientes con insuficiencia hepática leve (TB 1,0 a 1,5 × ULN o AST > ULN, según se define usando los criterios del Instituto Nacional del Cáncer de disfunción hepática; n=76) en comparación con pacientes con función hepática normal (TB y AST ≤ULN; n=708) en los análisis farmacocinéticos poblacionales. No se hallaron diferencias clínicamente importantes en el clearance del ipilimumab entre pacientes con insuficiencia hepática leve y aquellos con función hepática normal. YERVOY no ha sido estudiado en pacientes con insuficiencia hepática moderada (TB > 1,5 a 3 × ULN y cualquier valor de AST) o severa (TB > 3 × ULN y cualquier valor de AST). Toxicología no clínica: Carcinogénesis, mutagénesis, disfunción de la fertilidad: Carcinogénesis: El potencial carcinogénico del ipilimumab no se ha evaluado en estudios en animales a largo plazo. Mutagénesis: El potencial genotóxico del ipilimumab no se ha evaluado. Disfunción de la fertilidad: No se han llevado a cabo estudios de la fertilidad con ipilimumab. Toxicología y/o farmacología en animales: Además de severos resultados de aborto, nacimiento sin vida y muerte postnatal observados en ejemplares de Macaca fascicularis preñadas que recibieron ipilimumab cada 3 semanas desde el inicio de la organogénesis en el primer trimestre hasta el parto, se identificaron anormalidades en el desarrollo del sistema urogenital de 2 monos infantes expuestos in utero a 30 mg/kg de ipilimumab (7,2 veces el AUC en humanos con la dosis clínicamente recomendada). Un mono infante hembra presentó agenesia renal unilateral en el riñón y uréter izquierdos, y 1 mono infante macho presentó uretra imperforada con obstrucción urinaria y edema escrotal subcutáneo asociados. Los ratones heterocigotas, CTLA-4 (CTLA-4+/-), modificados por ingeniería genética, el objetivo del ipilimumab, parecieron sanos, y sus crías heterocigóticas, CTLA-4+/-, también nacieron sanas. Los ratones heterocigotas con CTLA-4+/- que se aparearon tuvieron crías con CTLA-4 deficiente (ratones homocigotas negativos, CTLA-4-/-). Las crías de ratones homocigotas, CTLA-4-/-, parecieron sanas al momento del nacimiento, mostraron signos de enfermedad linfoproliferativa multiorgánica a las 2 semanas del nacimiento, y todas murieron a las 3-4 semanas del nacimiento con linfoproliferación masiva y destrucción multiorgánica de tejidos.
Indicaciones.
YERVOY (ipilimumab) está indicado para el tratamiento del melanoma metastásico o no extirpable.
Dosificación.
Dosis recomendada: La dosis recomendada de YERVOY es 3 mg/kg administrados en forma intravenosa durante 90 minutos, cada 3 semanas, hasta un total de cuatro dosis. Modificaciones de la dosis recomendada: Interrumpir la dosis programada de YERVOY si se presentan reacciones adversas moderadas mediadas por la respuesta inmunitaria o una endocrinopatía sintomática. En el caso de los pacientes en quienes las reacciones adversas han desaparecido en forma parcial o completa (grados 0-1) y que reciben menos de 7,5 mg de prednisona o una sustancia equivalente al día, continuar con la administración de YERVOY en dosis de 3 mg/kg cada 3 semanas hasta que se administren las 4 dosis programadas o hasta que hayan transcurrido 16 semanas a partir de la primera dosis, lo que ocurra en primer término. Discontinuar la administración de YERVOY en forma permanente si: 1. Se presentan reacciones adversas moderadas persistentes o no se puede reducir la dosis de corticoesteroides a 7,5 mg de prednisona o una sustancia equivalente al día. 2. No se puede completar el tratamiento en forma completa dentro de las 16 semanas a partir de la administración de la primera dosis. 3. Se presentan reacciones adversas graves o con riesgo de muerte, lo cual incluye cualquiera de las siguientes afecciones: A. Colitis con dolor abdominal; fiebre; íleo o signos peritoneales; aumento de la frecuencia de deposiciones (7 o más sobre el nivel basal); incontinencia fecal; necesidad de hidratación intravenosa durante más de 24 horas; hemorragia gastrointestinal y perforación gastrointestinal. B. Niveles de aspartato aminotransferasa (AST) o de alanina aminotransferasa (ALT) > 5 veces el límite superior normal o niveles de bilirrubina total > 3 veces el límite superior normal. C. Síndrome de Stevens-Johnson, necrólisis epidérmica tóxica o erupción cutánea complicada por úlceras dérmicas en todo su espesor o manifestaciones necróticas, ampollosas o hemorrágicas. D. Neuropatía motora o sensorial grave, síndrome de Guillain-Barré o miastenia gravis. E. Reacciones graves mediadas por la respuesta inmunitaria que afecten cualquier sistema orgánico (p. ej., nefritis, neumonitis, pancreatitis, miocarditis no infecciosa). F. Enfermedad ocular mediada por la respuesta inmunitaria que no responde al tratamiento inmunosupresor tópico. Preparación y administración: No agitar el producto. Verificar visualmente que no haya partículas ni decoloración en los productos farmacológicos parenterales antes de la administración. Desechar el vial si la solución está turbia, presenta una decoloración pronunciada (el color de la solución puede ser amarillo pálido) o si presenta partículas extrañas que no sean transparentes o blancas, o partículas amorfas. Preparación de la solución: 1. Dejar reposar los viales a temperatura ambiente durante, aproximadamente, 5 minutos antes de preparar la infusión. 2. Extraer el volumen necesario de YERVOY y colocarlo en una bolsa para infusión intravenosa. 3. Diluir con cloruro de sodio para inyección al 0,9% o dextrosa al 5% para inyección, a fin de preparar una solución diluida con una concentración final que oscile entre 1 mg/mL a 2 mg/mL. Mezclar la solución diluida por inversión suave. 4. Almacenar la solución diluida durante no más de 24 horas bajo refrigeración 2°C a 8°C) o a una temperatura entre 20°C a 25°C. 5. Descartar la porción de solución no utilizada. Descartar los viales parcialmente usados o vacíos de YERVOY. Instrucciones de administración: 1. No mezclar YERVOY con otros productos medicinales ni administrarlo como infusión junto con otros productos medicinales. 2. Limpiar la vía intravenosa con cloruro de sodio para inyección al 0,9% o dextrosa para inyección al 5%, después de cada dosis. 3. Administrar la solución diluida durante 90 minutos a través de una vía intravenosa que contenga un filtro en línea estéril, no pirogénico, con baja unión a proteínas. 4. Administrar la solución diluida dentro de las 24 horas de su preparación. Formas de dosificación y concentraciones: YERVOY (ipilimumab) Solución Inyectable para Infusión Intravenosa: En frasco ampolla/vial de 50 mg/10mL (5 mg/mL). En frasco ampolla/vial de 200 mg/40 mL (5 mg/mL).
Contraindicaciones.
YERVOY está contraindicado en pacientes con hipersensibilidad previamente demostrada a ipilimumab o a cualquier componente del producto.
Reacciones adversas.
Las siguientes reacciones adversas se analizan en mayor detalle en otras secciones del prospecto. Enterocolitis mediada por la respuesta inmunitaria. Hepatitis mediada por la respuesta inmunitaria. Dermatitis mediada por la respuesta inmunitaria. Neuropatías mediadas por la respuesta inmunitaria. Endocrinopatías mediadas por la respuesta inmunitaria. Otras reacciones adversas mediadas por la respuesta inmunitaria, incluidas las manifestaciones oculares. Dado que los estudios clínicos se llevan a cabo en condiciones muy diferentes, los índices de reacciones adversas observados no pueden compararse en forma directa con los índices de otros estudios clínicos ni con la experiencia obtenida con tratamientos de la misma clase, y pueden no reflejar los índices observados en la práctica clínica. El programa de desarrollo clínico excluyó a los pacientes que presentaban enfermedades activas autoinmunitarias o a los que recibían inmunosupresores sistémicos en caso de trasplante de órgano. Se evaluó la exposición a 3 mg/kg de YERVOY en 4 dosis administradas mediante infusión intravenosa en pacientes con melanoma metastásico o no extirpable, previamente tratados, en un estudio clínico, aleatorizado, doble ciego (Estudio 1). Ciento treinta y un pacientes (mediana de edad: 57 años, 60% hombres) recibieron YERVOY como agente único, 380 pacientes (mediana de edad: 56 años, 61% hombres) recibieron YERVOY junto con una vacuna en investigación de péptidos gp100 (gp100), y 132 pacientes (mediana de edad: 57 años, 54% hombres) recibieron la vacuna de péptidos gp100 únicamente. Los pacientes del estudio recibieron una mediana de 4 dosis (entre 1 y 4 dosis). Se discontinuó la administración de YERVOY a causa de las reacciones adversas en el 10% de los pacientes. Las reacciones adversas más frecuentes (≥5%) que se presentaron en los pacientes que recibieron 3 mg/kg de YERVOY fueron: fatiga, diarrea, prurito, erupción cutánea y colitis. La Tabla 1 incluye determinadas reacciones adversas observadas en el Estudio 1, que se presentaron en, al menos, el 5% de los pacientes en los grupos que recibían YERVOY y que registraron un aumento de, al menos, el 5% en su incidencia en comparación con el grupo de control que recibía gp100, para los eventos de todos los grados, y un aumento en la incidencia de, al menos, el 1% con respecto al grupo de control para los eventos de grados 3-5.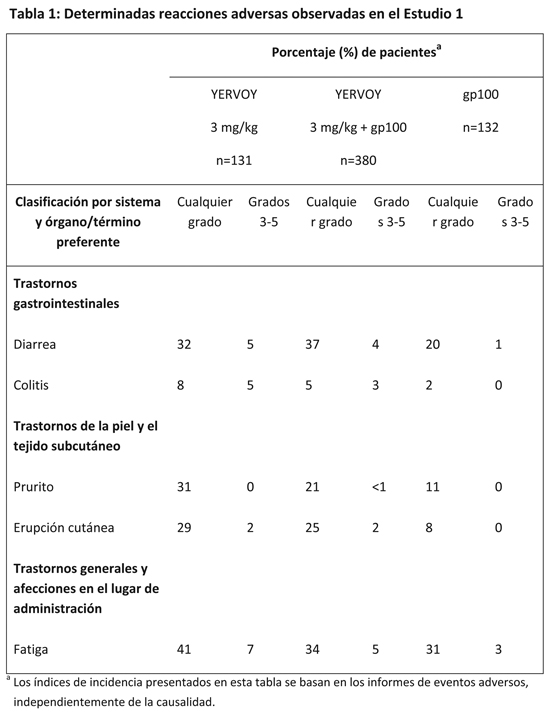
La Tabla 2 incluye la incidencia por paciente de reacciones adversas graves, con riesgo de muerte o mortales mediadas por la respuesta inmunitaria obtenida a partir del Estudio 1.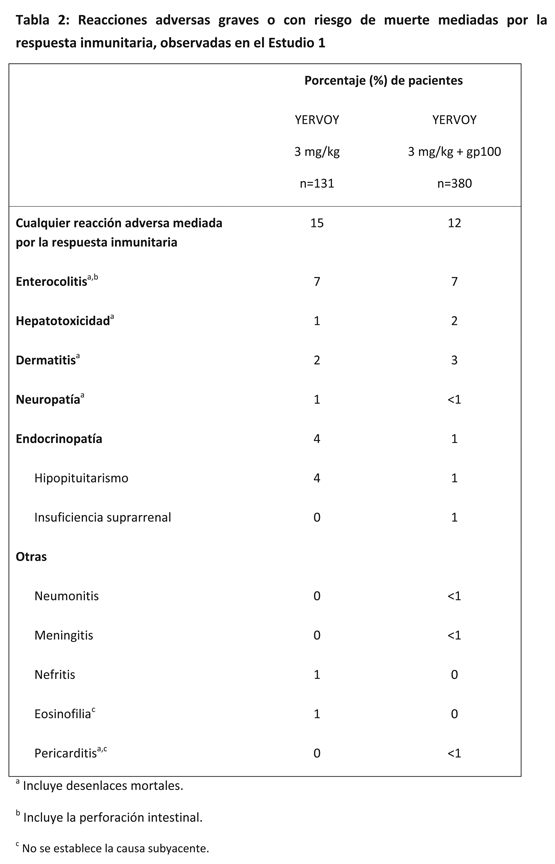
En los estudios clínicos en los que se utilizaron dosis de YERVOY de entre 0,3 a 10 mg/kg, también se informaron las siguientes reacciones adversas (incidencia menor del 1%, a menos que se especifique lo contrario): urticaria (2%), úlcera del intestino grueso, esofagitis, síndrome disneico agudo, insuficiencia renal, reacción a la infusión, sarcoidosis, hipoacusia neurosensorial, neuropatía central autoinmune (encefalitis), miositis, polimiositis y miositis ocular. En función de la experiencia obtenida en todo el programa clínico del melanoma, la incidencia y la gravedad de la enterocolitis y la hepatitis parecen estar relacionadas con la dosis. Inmunogenicidad: En los estudios clínicos, el 1,1% de los 1.024 pacientes evaluables tuvieron un resultado positivo en un ensayo de electroquimioluminiscencia (ECL) para los anticuerpos de unión contra el ipilimumab. Este ensayo presenta limitaciones sustanciales para detectar los anticuerpos contra el ipilimumab en presencia del ipilimumab. No se observaron reacciones peri-infusionales ni reacciones relacionadas con la infusión en relación con la hipersensibilidad o la anafilaxia en estos 11 pacientes, tampoco se detectaron anticuerpos neutralizantes contra el ipilimumab. Dado que los niveles mínimos del ipilimumab interfieren en los resultados del ensayo de ECL, se realizó un análisis de un subgrupo en la cohorte de dosis con los niveles mínimos. En este análisis, el 6,9% de los 58 pacientes evaluables que recibieron tratamiento con dosis de 0,3 mg/kg tuvieron un resultado positivo en la prueba de anticuerpos de unión contra el ipilimumab. Los resultados del ensayo de inmunogenicidad dependen en gran medida de varios factores, que incluyen: la sensibilidad y especificidad del ensayo, la metodología del ensayo, la manipulación de las muestras, el momento en que se extraen las muestras, los medicamentos concomitantes y las enfermedades subyacentes. Por estos motivos, la comparación de la incidencia de los anticuerpos contra YERVOY con las incidencias de los anticuerpos contra otros productos puede ser engañosa. Advertencias. Enterocolitis mediada por la respuesta inmunitaria: En el Estudio 1, hubo 34 casos (7%) de enterocolitis mediada por la respuesta inmunitaria en pacientes que fueron tratados con YERVOY. Estos casos fueron graves, con riesgo de muerte o mortales (diarrea con 7 o más deposiciones sobre el nivel basal, fiebre, íleo y signos peritoneales; grados 3-5). Asimismo, 28 pacientes (5%) que recibían tratamiento con YERVOY presentaron enterocolitis moderada (diarrea con hasta 6 deposiciones sobre el nivel basal, dolor abdominal, moco o sangre en las deposiciones; grado 2). Entre todos los pacientes que recibieron tratamiento con YERVOY (n=511), 5 (1%) pacientes desarrollaron perforación intestinal, 4 (0,8%) pacientes murieron a causa de complicaciones, y 26 (5%) pacientes fueron hospitalizados a causa de una enterocolitis grave. La mediana del tiempo hasta el inicio de estas reacciones fue 7,4 semanas (rango de 1,6 a 13,4) y 6,3 semanas (rango de 0,3 a 18,9) en los pacientes con enterocolitis de grados 3-5 y en pacientes con enterocolitis de grado 2, respectivamente, luego de haber comenzado la administración de YERVOY. Veintinueve pacientes (85%) con enterocolitis de grados 3-5 recibieron tratamiento con dosis altas de corticoesteroides (≥40 mg de prednisona o un equivalente al día), con una dosis media de 80 mg/día de prednisona o un equivalente; la mediana de duración del tratamiento fue de 2,3 semanas (se extendió hasta 13,9 semanas) y luego la administración de corticoesteroides se redujo progresivamente. Entre los 28 pacientes con enterocolitis moderada, el 46% no recibió corticoesteroides sistémicos, el 29% recibió tratamiento con < 40 mg de prednisona o una sustancia equivalente al día durante una mediana de duración de 5,1 semanas, y el 25% de los pacientes fueron tratados con dosis altas de corticoesteroides durante una mediana de tiempo de 10 días antes de reducir progresivamente la administración de corticoesteroides. Se administró infliximab a 5 de los 62 pacientes (8%) que presentaron enterocolitis moderada, grave o con riesgo de muerte, mediada por la respuesta inmunitaria luego de una respuesta inadecuada a los corticoesteroides. De los 34 pacientes con enterocolitis de grados 3-5, el 74% resolvió en forma completa, el 3% presentó una mejoría a grado 2, y el 24% no presentó mejoría. Entre los 28 pacientes con enterocolitis de grado 2, el 79% presentó una mejoría completa, el 11% presentó una mejoría, y el 11% no presentó mejoría. Se debe controlar a los pacientes para detectar signos y síntomas de enterocolitis (como diarrea, dolor abdominal, moco o sangre en las heces, con o sin fiebre) y de perforación intestinal (como íleo y signos peritoneales). En los pacientes sintomáticos, se deben descartar las causas infecciosas y se debe considerar una evaluación endoscópica en el caso de los síntomas persistentes o graves. Discontinuar la administración de YERVOY en forma permanente en los pacientes que presentan enterocolitis grave y comenzar el tratamiento con corticoesteroides sistémicos a una dosis de 1 a 2 mg/kg/día de prednisona o una sustancia equivalente. Una vez que el paciente mejore y presente el grado 1 o un grado menor, se debe comenzar a disminuir progresivamente la administración de corticoesteroides y continuar disminuyendo esta administración durante, al menos, 1 mes. En estudios clínicos, la disminución progresiva de la administración de corticoesteroides provocó, en algunos pacientes, la recurrencia o el empeoramiento de los síntomas de la enterocolitis. Se debe interrumpir el tratamiento con YERVOY si el paciente presenta enterocolitis moderada; se debe administrar un tratamiento antidiarreico y, si la afección continúa durante más de 1 semana, se debe iniciar un tratamiento con corticoesteroides sistémicos a una dosis de 0,5 mg/kg/día de prednisona o una sustancia equivalente. Hepatitis mediada por la respuesta inmunitaria: En el Estudio 1, 8 (2%) pacientes tratados con YERVOY presentaron una hepatotoxicidad grave, con riesgo de muerte o mortal (elevaciones de la AST o la ALT 5 veces superiores al límite superior normal o elevaciones en los niveles de bilirrubina total 3 veces superiores al límite superior normal; grados 3-5). El 0,2% de los pacientes que recibieron tratamiento con YERVOY presentaron una insuficiencia hepática mortal, y el 0,4% de los pacientes tuvieron que ser hospitalizados. Otros 13 pacientes (2,5%) presentaron una hepatotoxicidad moderada que se manifestó en anomalías en las pruebas de la función hepática (elevaciones de la AST o la ALT de más de 2,5 veces, aunque no más de 5, del límite superior normal, o una elevación de la bilirrubina total de más de 1,5 veces, aunque no más de 3, del límite superior normal; grado 2). No se confirmó patología subyacente en todos los pacientes, sin embargo en algunos casos la biopsia confirmó la hepatitis mediada por la respuesta inmunitaria. No hubo una cantidad suficiente de pacientes cuya hepatitis haya sido confirmada mediante biopsia, a fin de caracterizar la evolución clínica de este evento. Se deben controlar las pruebas de la función hepática (niveles de transaminasa hepática y bilirrubina), y se debe evaluar a los pacientes para detectar signos y síntomas de hepatotoxicidad antes de cada dosis de YERVOY. En los pacientes que presentan hepatotoxicidad, se deben descartar causas infecciosas o malignas, y se debe aumentar la frecuencia de las pruebas de la función hepática hasta su resolución. Se debe discontinuar la administración de YERVOY de modo permanente en los pacientes con hepatotoxicidad de grados 3-5, y se debe administrar un tratamiento con corticoesteroides sistémicos a una dosis de 1 a 2 mg/kg/día de prednisona o una sustancia equivalente. Cuando las pruebas de función hepática muestren una mejoría sostenida o los valores regresen al nivel basal, se debe comenzar a reducir en forma progresiva la administración de corticoesteroides durante 1 mes. En el programa de desarrollo clínico de YERVOY, se administró el tratamiento con micofenolato en los pacientes que presentaban hepatitis grave persistente a pesar de las dosis altas de corticoesteroides. Se debe interrumpir la administración de YERVOY en los pacientes que presentan una hepatotoxicidad de grado 2. Dermatitis mediada por la respuesta inmunitaria: En el Estudio 1, 13 (2,5%) pacientes tratados con YERVOY presentaron dermatitis grave, con riesgo de muerte o mortal mediada por la respuesta inmunitaria (p. ej., el síndrome de Stevens-Johnson, necrólisis epidérmica tóxica o erupción cutánea complicada por úlceras dérmicas de espesor total, o manifestaciones necróticas, ampollosas o hemorrágicas; grados 3-5). Un (0,2%) paciente murió como consecuencia de una necrólisis epidérmica tóxica, y otro paciente debió ser hospitalizado a causa de una dermatitis grave. Sesenta y tres (12%) pacientes presentaron dermatitis moderada (grado 2). La mediana del tiempo hasta el inicio de la dermatitis moderada, grave o con riesgo de muerte, mediada por la respuesta inmunitaria, fue de 3,1 semanas y se extendió hasta 17,3 semanas a partir del inicio de la administración de YERVOY. Siete (54%) pacientes tratados con YERVOY que presentaron dermatitis grave recibieron dosis altas de corticoesteroides (mediana de la dosis de 60 mg de prednisona/día o una sustancia equivalente) durante 14,9 semanas, como máximo, seguido por la disminución gradual de la administración de corticoesteroides. De estos 7 pacientes, 6 tuvieron una resolución completa; el tiempo hasta la mejoría completa fue de hasta 15,6 semanas. Entre estos 63 pacientes que presentaban dermatitis moderada, 25 (40%) recibieron tratamiento con corticoesteroides sistémicos (mediana de 60 mg/día de prednisona o una sustancia equivalente) durante una mediana de 2,1 semanas; 7 (11%) pacientes recibieron tratamiento únicamente con corticoesteroides tópicos, y 31 (49%) pacientes no recibieron ni corticoesteroides sistémicos ni tópicos. Se informó que 44 (70%) pacientes que presentaron dermatitis moderada tuvieron una mejoría completa, 7 (11%) mejoraron y presentaron una intensidad leve (grado 1), y 12 (19%) no informaron mejorías. Se debe controlar a los pacientes para detectar signos y síntomas de dermatitis, como erupción cutánea y prurito. A menos que se haya establecido otra causa, los signos y síntomas de la dermatitis deben considerarse mediados por la respuesta inmunitaria. Se debe discontinuar la administración de YERVOY de modo permanente en los pacientes que presentan el síndrome de Stevens-Johnson, necrólisis epidérmica tóxica o erupción cutánea complicada por úlceras dérmicas de espesor total, o manifestaciones necróticas, ampollosas o hemorrágicas. Se deben administrar corticoesteroides sistémicos a una dosis de 1 a 2 mg/kg/día de prednisona o una sustancia equivalente. Cuando la dermatitis está controlada, debe comenzar a disminuirse la administración de corticoesteroides en forma progresiva durante un período de, al menos, 1 mes. Se debe interrumpir la administración de YERVOY en pacientes que presentan signos y síntomas de moderados a graves. La dermatitis de leve a moderada, como la erupción cutánea o el prurito localizados, debe tratarse de modo sintomático. Se deben administrar corticoesteroides sistémicos o tópicos si no se observan mejorías en los síntomas en el término de 1 semana. Neuropatías mediadas por la respuesta inmunitaria: En el Estudio 1, se informaron 1 caso de síndrome de Guillain-Barré que resultó mortal y 1 caso de neuropatía motora periférica grave (grado 3). En el programa de desarrollo clínico de YERVOY, se han informado casos de miastenia gravis y otros casos de síndrome de Guillain-Barré. Se debe controlar a los pacientes para detectar síntomas de neuropatía motora o sensorial, como debilidad unilateral o bilateral, alteraciones sensoriales o parestesia. Se debe discontinuar la administración de YERVOY en forma permanente en los pacientes que presentan neuropatía grave (que interfiere en sus actividades diarias), como síndromes parecidos al de Guillain-Barré. Se debe administrar un tratamiento, según corresponda, para la neuropatía grave. Debe considerarse comenzar a administrar corticoesteroides sistémicos a una dosis de 1 a 2 mg/kg/día de prednisona o una sustancia equivalente para el tratamiento de la neuropatía grave. Se debe interrumpir la dosis de YERVOY en los pacientes que presentan neuropatía moderada (que no interfiere en sus actividades diarias). Endocrinopatías mediadas por la respuesta inmunitaria: En el Estudio 1, 9 (1,8%) pacientes tratados con YERVOY presentaron endocrinopatías graves o con riesgo de muerte mediadas por la respuesta inmunitaria (que requirieron hospitalización, tratamiento médico urgente o que interfirieron en sus actividades diarias; grados 3-4). Estos 9 pacientes presentaron hipopituitarismo, y algunos tuvieron algunas endocrinopatías concomitantes adicionales, como insuficiencia suprarrenal, hipogonadismo e hipotiroidismo. Seis de los nueve pacientes fueron hospitalizados a causa de endocrinopatías graves. Doce (2,3%) pacientes presentaron endocrinopatías moderadas (que requirieron reemplazo hormonal o tratamiento médico; grado 2). Estas endocrinopatías incluyeron: hipotiroidismo, insuficiencia suprarrenal, hipopituitarismo y 1 caso de hipertiroidismo y 1 caso de síndrome de Cushing. La mediana del tiempo hasta el inicio de la endocrinopatía de moderada a grave mediada por la respuesta inmunitaria fue de 11 semanas y se extendió hasta 19,3 semanas después de haber comenzado el tratamiento con YERVOY. De los 21 pacientes con endocrinopatía moderada o con riesgo de muerte, 17 pacientes necesitaron un tratamiento de reemplazo hormonal a largo plazo que incluyó, en la mayoría de los casos, hormonas suprarrenales (n=10) y hormonas tiroideas (n=13). Se debe controlar a los pacientes para detectar síntomas y signos clínicos de inflamación de la glándula pituitaria, insuficiencia suprarrenal (incluida la insuficiencia suprarrenal aguda), e hipertiroidismo o hipotiroidismo. Los pacientes pueden presentar fatiga, dolor de cabeza, cambios en el estado mental, dolor abdominal, hábitos intestinales inusuales e hipotensión, o síntomas no específicos que pueden parecerse a síntomas de otras afecciones, como metástasis en el cerebro o enfermedades subyacentes. A menos que se haya identificado otra causa, los signos y síntomas de las endocrinopatías deben considerarse mediados por la respuesta inmunitaria. Se deben realizar pruebas para controlar la función tiroidea y análisis bioquímicos clínicos al comienzo del tratamiento, antes de cada dosis, y según la indicación clínica en función de los síntomas. En una cantidad limitada de pacientes, se diagnosticó la inflamación de la glándula pituitaria mediante estudios de diagnóstico por imágenes a través del agrandamiento de la glándula pituitaria. Se debe interrumpir la administración de YERVOY en los pacientes sintomáticos. Se debe comenzar la administración de corticoesteroides sistémicos a una dosis de 1 a 2 mg/kg/día de prednisona o una sustancia equivalente, y comenzar un tratamiento adecuado de reemplazo hormonal. Otras reacciones adversas mediadas por la respuesta inmunitaria, incluidas las manifestaciones oculares: Se observaron las siguientes reacciones adversas clínicamente significativas mediadas por la respuesta inmunitaria en menos del 1% de los pacientes tratados con YERVOY en el Estudio 1: nefritis, neumonitis, meningitis, pericarditis, uveítis, iritis y anemia hemolítica. En el programa de desarrollo clínico de YERVOY, también se informaron las siguientes reacciones adversas probablemente mediadas por la respuesta inmunitaria con menos del 1% de incidencia: miocarditis, angiopatía, arteritis temporal, vasculitis, polimialgia reumática, conjuntivitis, blefaritis, epiescleritis, escleritis, vasculitis leucocitoclástica, eritema multiforme, psoriasis, pancreatitis, artritis y tiroiditis autoinmunitaria. Se debe discontinuar la administración de YERVOY en forma permanente si se presentan reacciones adversas clínicamente significativas o reacciones adversas graves mediadas por la respuesta inmunitaria. Se debe iniciar la administración de corticoesteroides sistémicos a una dosis de 1 a 2 mg/kg/día de prednisona o una sustancia equivalente para el tratamiento de las reacciones adversas graves mediadas por la respuesta inmunitaria. Se deben administrar gotas oftálmicas con corticoesteroides a los pacientes que desarrollan uveítis, iritis o epiescleritis. Se debe discontinuar la administración de YERVOY en forma permanente para el tratamiento de las enfermedades oculares mediadas por la respuesta inmunitaria que no responden al tratamiento inmunosupresor local. Reacciones adversas medidas por la respuesta inmunitaria: YERVOY puede provocar reacciones adversas graves y mortales mediadas por la respuesta inmunitaria a causa de la activación y proliferación de las células T.
Interacciones.
No se han realizado estudios formales de interacción medicamentosa con YERVOY. Uso en poblaciones específicas: Embarazo: Embarazo Categoría C: No se dispone de estudios adecuados y bien controlados sobre la administración de YERVOY en mujeres embarazadas. Se debe usar YERVOY durante el embarazo únicamente si el beneficio potencial justifica el riesgo potencial para el feto. En un estudio combinado sobre el desarrollo embrio-fetal, perinatal y postnatal, se administró ipilimumab a ejemplares preñadas de Macaca fascicularis cada 3 semanas a partir del inicio de la organogénesis en el primer trimestre hasta el parto, a niveles de exposición que eran 2,6 o 7,2 veces mayores por AUC que las exposiciones alcanzadas con la dosis clínica de 3 mg/kg de ipilimumab. No se detectaron efectos adversos relacionados con el tratamiento sobre la reproducción durante los primeros dos trimestres del embarazo. A partir del tercer trimestre, los grupos tratados con ipilimumab experimentaron mayor incidencia de toxicidades severas, incluidos abortos, nacimientos de crías muertas, partos prematuros (con el correspondiente menor peso al nacimiento) y mayor incidencia de mortalidad infantil de manera relacionada con la dosis, en comparación con los controles. La IgG1 humana atraviesa la barrera placentaria. Ipilimumab es una IgG1; por lo tanto, la madre puede transmitir ipilimumab al feto en desarrollo. Mujeres en período de lactancia: Se desconoce si ipilimumab se secreta a través de la leche humana. En las monas tratadas a niveles de dosis que dan como resultado exposiciones 2,6 y 7,2 veces mayores que aquellas alcanzadas en humanos con la dosis recomendada, el ipilimumab estuvo presente en la leche en concentraciones de 0,1 y 0,4 mcg/mL, lo cual representa una proporción de hasta 0,3% de la concentración sérica del fármaco. Debido a que muchos fármacos se secretan en la leche humana y al potencial de que se produzcan reacciones adversas graves en los niños lactantes a causa de la administración de YERVOY, debe tomarse una decisión respecto de interrumpir la lactancia o la administración de YERVOY considerándose la importancia de YERVOY para la madre. Uso pediátrico: No se han establecido la seguridad ni la eficacia de YERVOY en pacientes pediátricos. Uso geriátrico: De los 511 pacientes que recibieron tratamiento con YERVOY en dosis de 3 mg/kg, el 28% tenían 65 años o más. No se observaron diferencias generales en la eficacia ni en la seguridad entre los pacientes de edad avanzada (65 años o más) y los pacientes más jóvenes (menores de 65 años). Insuficiencia renal: No se han realizado estudios formales con YERVOY en pacientes con insuficiencia renal. Insuficiencia hepática: No se han llevado a cabo estudios formales de YERVOY en pacientes con insuficiencia hepática.
Conservación.
Almacenar YERVOY bajo refrigeración a una temperatura de 2°C a 8°C. No se debe congelar o agitar. Los frascos ampolla/viales deben protegerse de la luz.
Sobredosificación.
No se dispone de información acerca de la sobredosis con YERVOY.
Presentación.
YERVOY se encuentra disponible en las siguientes presentaciones: Un frasco ampolla/vial inyectable de 50 mg (5 mg/mL), de un solo uso. Un frasco ampolla/vial inyectable de 200 mg (5 mg/mL), de un solo uso.