ZELBORAF®
ROCHE
El vemurafenib es una molécula de bajo peso molecular que inhibe selectivamente las serina-treonincinasas BRAF oncogénicas. Código ATC: L01XE15.
Composición.
Principio activo: vemurafenib. Cada comprimido recubierto contiene 240 mg de vemurafenib (como coprecipitado de vemurafenib y succinato acetato de hipromelosa). Excipientes: dióxido de silicio coloidal anhidro, croscarmelosa sódica, hiprolosa, estearato de magnesio, alcohol polivinílico, dióxido de titanio, macrogol 3350, talco, óxido de hierro rojo. Forma farmacéutica: Comprimidos recubiertos. Vía de administración: Oral. Declaración de esterilidad / radiactividad: No procede.
Farmacología.
Propiedades farmacodinámicas: Mecanismo de acción: El vemurafenib es un inhibidor oral de bajo peso molecular de la forma activada de la enzima serina-treonincinasa BRAF. Mutaciones en el gen BRAF dan lugar a una activación constitutiva de la proteína BRAF, la cual puede fomentar una señalización y proliferación celular excesiva en ausencia de factores de crecimiento típicos. Como inhibidor potente y selectivo de la proteína oncogénica BRAF, el vemurafenib suprime la señalización cascada abajo a través de la vía dependiente de la proteincinasa de activación mitogénica (MAPK). El sustrato de BRAF mejor caracterizado es MEK. La fosforilación de MEK por BRAF conduce a la activación de pMEK. Por su parte, pMEK fosforila ERK a pERK, el cual se transloca al núcleo donde activa factores de transcripción responsables de estimular la proliferación y la supervivencia celular. Estudios preclínicos in vitro han demostrado que el vemurafenib es un potente inhibidor de la fosforilación y la activación de MEK y ERK y, por consiguiente, suprime la proliferación celular en las células tumorales que expresan proteínas con la mutación BRAFV600 (ver tabla siguiente).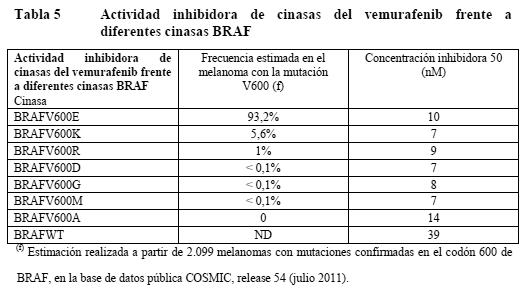
Ensayos clínicos / Eficacia: La eficacia del vemurafenib se ha evaluado en 675 pacientes de un estudio clínico de fase III y 132 pacientes de un estudio de fase II. Antes de entrar los pacientes en el estudio, se realizó una prueba diagnóstica de la presencia de mutaciones BRAF V600 utilizando el Cobas® 4800 BRAF V600 Mutation Test. Pacientes no tratados previamente: Los resultados de un estudio de fase III abierto, aleatorizado, multicéntrico e internacional respaldan el uso del vemurafenib en pacientes con melanoma metastásico o irresecable positivo para la mutación BRAFV600 que no habían sido tratados previamente. Se aleatorizó a los pacientes al tratamiento con vemurafenib (960 mg dos veces al día) o dacarbazina (1.000 mg/m2 cada 3 semanas). Un total de 336 pacientes fueron asignados aleatorizadamente a un grupo del vemurafenib (n=337) o un grupo de la dacarbazina (n=338). La aleatorización se estratificó según el estadio de la enfermedad, la concentración sérica de LDH, el estado general según la escala de ECOG y la región geográfica. Las características basales de los grupos estaban bien equilibradas. De los pacientes aleatorizados al grupo del vemurafenib, la mayoría eran varones (59%) y de origen caucásico (99%), la mediana de la edad era de 56 años (28% tenían ≥ 65 años), todos los pacientes presentaban un estado general según la escala de ECOG de 0 o 1 y la mayoría se hallaba en el estadio M1c de la enfermedad (66%). Las variables principales de la eficacia de este estudio eran la supervivencia global (SG) y la supervivencia sin progresión (SSP). Importantes variables secundarias de valoración eran la mejor tasa global de respuesta (MTGR) y la duración de la respuesta. Se observaron mejoras estadísticamente significativas y clínicamente importantes de las variables principales de valoración SG (p < 0,0001) y SSP (p < 0,0001) (prueba de rangos logarítmicos no estratificada). En el momento del informe a los tres meses habían fallecido 200 pacientes en total (78 del grupo de vemurafenib y 122 del de dacarbazina). La SG era más larga en el grupo del vemurafenib que en el de la dacarbazina, siendo la hazard ratio de 0,44 (IC del 95%: 0,33 - 0,59), lo que representa un descenso del riesgo de fallecer del 56% con el vemurafenib en comparación con la dacarbazina. La tasa de supervivencia a los 6 meses estimada por el método de Kaplan-Meier era del 83% (IC del 95%: 79% - 87%) con el vemurafenib y del 63% (IC del 95%: 57 - 69%) con la dacarbazina. En el momento del análisis, la mediana de la SG estimada por el método de Kaplan-Meier no se había alcanzado en el grupo del vemurafenib (IC del 95%: 9,6-no alcanzada) y de 7,9 meses en el de la dacarbazina (IC del 95%: 7,3 - 9,6). La SSP evaluada por el investigador fue mayor con el vemurafenib que con la dacarbazina, con una hazard ratio de progresión o muerte (SSP) de 0,27 (IC del 95%: 0,20 - 0,33), que representa un descenso del 74% del riesgo de progresión o muerte con el vemurafenib en comparación con la dacarbazina. Las estimaciones de Kaplan-Meier de la SSP a los 6 meses eran del 47% (IC del 95%: 38% - 55%) para el vemurafenib y del 12% (IC del 95%: 7% - 18%) para la dacarbazina. La variable secundaria de valoración mejor tasa global de respuesta (RC + RP) evaluada por el investigador mejoró significativamente (p < 0,0001) en el grupo del vemurafenib (48,4%) (IC del 95%: 41,6% - 55,2%) en comparación con el de dacarbazina (5,5%) (IC del 95%: 2,8% - 9,3%). En el 37% de los pacientes tratados con vemurafenib y el 24% de los que recibieron dacarbazina se estabilizó la enfermedad según los criterios RECIST 1.1. En todos los subgrupos (edad, sexo, LDH basal, estado general según la escala de ECOG, estadio de la enfermedad metastásica) y regiones geográficas se observaron mejoras de la SG, la SSP y mejor tasa global de respuesta confirmada a favor del vemurafenib. La mediana de seguimiento de la SG en el grupo del vemurafenib fue de 6,2 meses (intervalo: 0,4 - 13,9) y de 4,5 meses en el de la dacarbazina (intervalo: < 0,1 - 11,7). En las Figuras 1 y 2 se resumen los resultados relativos a la eficacia.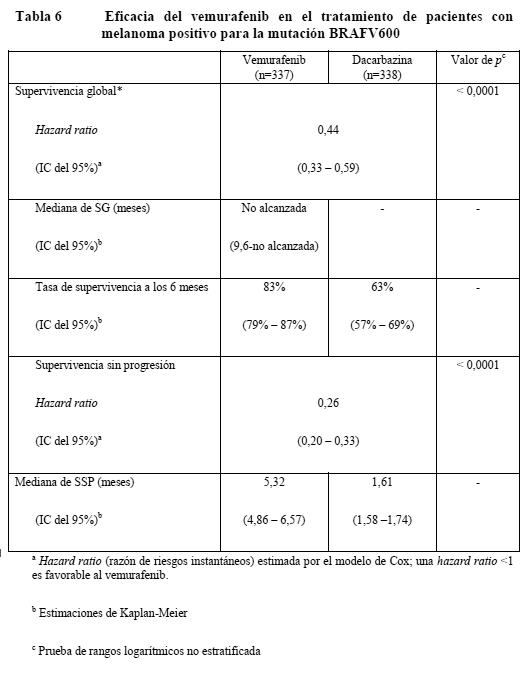
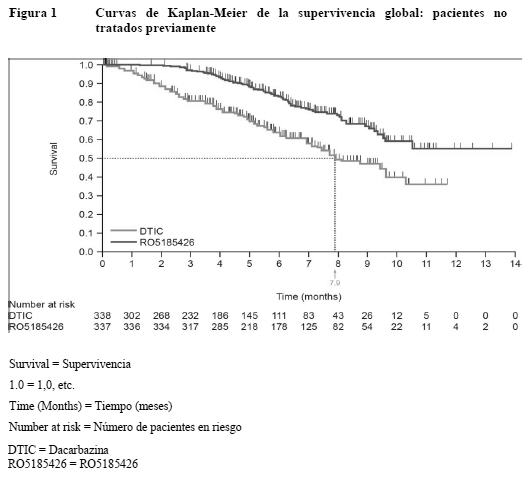
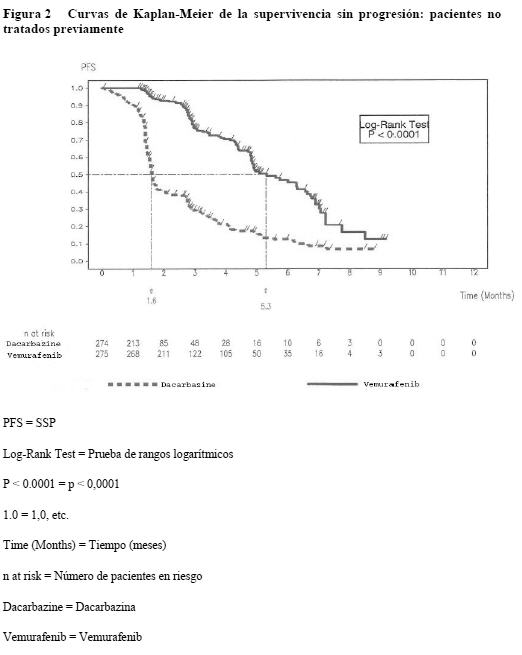
La proporción de pacientes que mejoraron según la evaluación del médico del estado general fue mayor en el grupo del vemurafenib (63,4%) (IC del 95%: 57% - 69%) que en el de la dacarbazina (20,2%) (IC del 95%: 15% - 26%). Pacientes que respondieron al menos a un tratamiento sistémico previo: Se realizó un estudio de fase II con un solo grupo, multicéntrico y multinacional en 132 pacientes con melanoma metastásico que habían recibido previamente al menos un tratamiento. La mediana de edad era de 52 años, con un 19% de los pacientes por encima de los 65 años. La mayoría eran varones (61%), de origen caucásico (99%) y se hallaban en el estadio M1c de la enfermedad (61%). El 49% de los pacientes no había respondido a ≥ 2 terapias previas. La mediana de seguimiento fue de 6,87 meses (intervalo: 0,6 - 11,3 meses). La variable principal de valoración tasa global de respuesta confirmada (RC + RP) evaluada por un CEIC (Comité ético de investigación clínica) independiente fue del 52% (IC del 95%: 43% - 61%). La mediana del tiempo hasta la respuesta fue de 1,4 mes, con el 75% de las respuestas al cabo de 1,6 mes de tratamiento. La mediana de la duración de la respuesta evaluada por un CEIC independiente fue de 6,5 meses (IC del 95%: 5,6-no alcanzado). En el 30% de los pacientes se observó enfermedad estable según los criterios RECIST 1.1. La mediana de supervivencia global no se ha alcanzado (IC del 95%: 9,5-no alcanzado), y la tasa de supervivencia a los 6 meses era del 77% (IC del 95%: 70% - 85%). La mediana de SSP era de 6,1 meses (IC del 95%: 5,5 - 6,9), y la tasa de SSP a los 6 meses era del 52% (IC del 95%: 43% - 61%). Propiedades farmacocinéticas: Los parámetros farmacocinéticos del vemurafenib se determinaron mediante un análisis no compartimental en un estudio de fase I y otro de fase III. Los valores medios de Cmáx, Cmín y ABC0-12 h fueron aproximadamente de 62 mg/ml, 53 mg/ml y 600 mg·h/ml, respectivamente. En un análisis de farmacocinética poblacional de los datos conjuntos de 458 pacientes, la mediana estimada de Cmáx, Cmín y ABC en equilibrio estacionario era de 62 mg/ml, 59 mg/ml y 734 mg·h/ml, respectivamente. La mediana del cociente de acumulación estimado para una pauta de dos tomas diarias es de 7,36. La farmacocinética del vemurafenib es proporcional a la dosis entre 240 mg y 960 mg dos veces al día, y el análisis de farmacocinética poblacional también confirmó que la farmacocinética del vemurafenib es lineal. Absorción: El vemurafenib tomado en una dosis de 960 mg dos veces al día en forma de comprimidos de 240 mg se absorbe con una mediana de Tmáx de aproximadamente 4 horas. Tomado el vemurafenib en dosis múltiples de 960 mg dos veces al día, se observa una marcada acumulación con una alta variabilidad interindividual. En el estudio de fase II, la concentración plasmática media de vemurafenib a las cuatro horas de la administración aumentó de 3,6 mg/ml el día 1 a 49,0 mg/ml el día 15 (intervalo: 5,4 - 118 mg/ml). Los alimentos (comidas ricas en grasa) elevan la biodisponibilidad relativa de una dosis única de 960 mg de vemurafenib. Los cocientes de la media geométrica entre el estado posprandial y en ayunas para la Cmáx y el ABB eran de 2,6 y 4,7, respectivamente. La mediana de Tmáx pasaba de 4 a 8 horas cuando una dosis única de vemurafenib se tomaba con alimentos. Los datos relativos a la seguridad y la eficacia en los estudios fundamentales procedían de pacientes que tomaron el vemurafenib con y sin alimentos. En equilibrio estacionario (alcanzado el día 15 en el 80% de los pacientes), la exposición plasmática media al vemurafenib permanece estable (concentración antes de la dosis matinal y a las 2 - 4 horas) según refleja la relación media de 1,13. Una marcada variabilidad interindividual similar en la exposición plasmática se observó en estado de equilibrio estacionario independientemente de la reducción de la dosis. Se estima que, tras la administración oral, la constante de la tasa de absorción en los pacientes con melanoma metastásico es de 0,19 h-1 (con una variabilidad interindividual del 101%). Distribución: El volumen de distribución poblacional estimado del vemurafenib en los pacientes con melanoma metastásico es de 91 litros (con una variabilidad interindividual del 64,8%). In vitro, el vemurafenib se une intensamente a las proteínas plasmáticas del ser humano ( > 99%). Metabolismo: Las proporciones relativas de vemurafenib y sus metabolitos se caracterizaron en un estudio de balance de masas en el ser humano, en estado de equilibrio, con una dosis única de vemurafenib marcado con C14 administrado por vía oral. En promedio, el 95% de la dosis se recuperó dentro de los 18 días siguientes a la administración. La mayor parte (94%) en las heces y < 1% en la orina. La principal enzima responsable del metabolismo del vemurafenib in vitro es CYP3A4, pero también se han identificado metabolitos de conjugación (glucuronización y glucosilación) en el ser humano. No obstante, el principal componente en plasma era el fármaco original (95%). Aunque el metabolismo no parece dar lugar a una cantidad importante de metabolitos en el plasma, no cabe excluir la importancia del metabolismo en la excreción. Eliminación: El aclaramiento aparente poblacional estimado del vemurafenib en los pacientes con melanoma metastásico es de 29,3 L (con una variabilidad interindividual del 31,9%). La mediana de la semivida de eliminación individual estimada del vemurafenib es de 56,9 horas (intervalo de los percentiles 5 y 95: 29,8 - 119,5 horas). Farmacocinética en poblaciones especiales: Ancianos: De acuerdo con el análisis de farmacocinética poblacional, la edad no tiene ningún efecto estadísticamente significativo en la farmacocinética del vemurafenib. Sexo: El análisis de farmacocinética poblacional puso de manifiesto que el sexo tiene una importancia estadísticamente significativa en la variabilidad interindividual, siendo en los varones el aclaramiento aparente (CL/F) un 17% mayor y el volumen aparente de distribución (V/F) un 48% mayor. Ahora bien, los resultados del análisis poblacional muestran que las diferencias en la exposición son relativamente pequeñas (con mediana estimada a las 12 horas en equilibrio estacionario de ABC y Cmáx de 792 mg·h/ml y 67 mg/ml en las mujeres y 696 mg·h/ml y 63 mg/ml en los varones, respectivamente), lo cual indica que no es necesario ajustar la dosis en función del sexo. Niños: No se han realizado estudios de farmacocinética del vemurafenib en niños. Insuficiencia renal: Según el análisis de farmacocinética poblacional con los datos de los ensayos clínicos en pacientes con melanoma metastásico, la insuficiencia renal leve y moderada no influía en el aclaramiento aparente del vemurafenib (aclaramiento de creatinina: > 30 ml/min). No hay datos clínicos y farmacocinéticos suficientes para determinar la necesidad potencial de ajustar la dosis en pacientes con insuficiencia renal grave (aclaramiento de creatinina: < 29 ml/min) (ver Pautas posológicas especiales e Insuficiencia renal). Insuficiencia hepática: De acuerdo con los datos preclínicos y los de un estudio de balance de masas en el ser humano, la mayor parte del vemurafenib se elimina por vía hepática. Según el análisis de farmacocinética poblacional con los datos de los ensayos clínicos en pacientes con melanoma metastásico, aumentos de AST, ALT y bilirrubina total hasta tres veces el límite superior de la normalidad no influían en el aclaramiento aparente del vemurafenib. No se puede determinar la necesidad potencial de ajustar la dosis en pacientes con insuficiencia hepática grave, ya que no hay datos clínicos y farmacocinéticos suficientes para determinar el efecto de un deterioro hepático metabólico o excretor en la farmacocinética del vemurafenib (ver Pautas posológicas especiales e Insuficiencia hepática). Datos preclínicos sobre seguridad: Carcinogenicidad: No se han realizado estudios de carcinogenicidad. Mutagenicidad: Todos los estudios habituales de genotoxicidad con el vemurafenib fueron negativos. Trastornos de la fecundidad: No se han realizado estudios preclínicos de fertilidad. En los estudios toxicológicos de dosis múltiples no se detectaron anomalías en los órganos reproductores. Teratogenicidad: El vemurafenib no reveló ningún signo de teratogenicidad en embriones/fetos de rata en dosis de hasta 250 mg/kg/día (una 1,7 vez la exposición clínica humana según el ABC) ni en embriones/fetos de conejo en dosis de hasta 450 mg/kg/día (una 0,7 vez la exposición clínica humana según el ABC). Los niveles de fármaco en los fetos eran un 3 - 5% más altos que en las madres, lo cual indica que el vemurafenib tiene el potencial para la transmisión de la madre al feto. No se han realizado estudios específicos del vemurafenib en animales para evaluar su efecto en la fertilidad. Ahora bien, en los estudios de dosis repetidas no se detectaron alteraciones histopatológicas en los órganos reproductores de machos y hembras con dosis de hasta 450 mg/kg/día en la rata (una 0,6 y 1,6 vez, respectivamente, la exposición humana según valores de ABC) y el perro (una 0,4 vez la exposición humana según valores de ABC tanto en los machos como en las hembras). Otros efectos: En estudios toxicológicos de dosis repetidas se identificaron el hígado y la médula ósea como órganos diana en el perro. En un estudio de 13 semanas en perros, con dos dosis diarias, se observaron efectos tóxicos reversibles en el hígado (necrosis y degeneración hepatocelular) con exposiciones por debajo de la exposición clínica (según comparaciones del ABC). En un estudio de 39 semanas en perros prematuramente terminado se detectó necrosis focal de la médula ósea en un perro con dos dosis diarias y una exposición dentro del intervalo de exposiciones clínicas. Se puso de manifiesto que el vemurafenib era fototóxico in vitro en fibroblastos cultivados de ratón tras una radiación UVA, pero no lo fue in vivo en un estudio en ratas. In vitro se observó inhibición del citocromo CYP2C9 (CI50: 5,9 mM).
Indicaciones.
Vemurafenib está indicado en monoterapia para el tratamiento de pacientes adultos con melanoma no resecable o metastásico con mutación de BRAF V600 positiva.
Dosificación.
El tratamiento con vemurafenib debe iniciarse y ser supervisado por un médico especializado en el uso de medicamentos anticancerígenos. Antes de comenzar el tratamiento con vemurafenib, los pacientes deben tener un diagnóstico de mutación BRAF V600 positiva en el tumor, confirmado por un test validado. Dosis habitual: La dosis recomendada de vemurafenib es de 960 mg (cuatro comprimidos de 240 mg) dos veces al día. La primera dosis debe tomarse por la mañana y la segunda por la tarde, aproximadamente unas 12 horas después. Cada dosis puede tomarse con o sin alimentos pero debe evitarse el consumo constante de las dos dosis diarias con el estómago vacío (ver Absorción). Los comprimidos de Zelboraf deben tomarse enteros, con un vaso de agua. Los comprimidos de Zelboraf no deben masticarse ni triturarse. Duración del tratamiento: Se recomienda mantener el tratamiento con Zelboraf hasta la progresión de la enfermedad o la aparición de toxicidad inaceptable (ver Tablas 1 y 2). Dosis no tomadas: En caso de saltarse una dosis, se puede tomar después hasta 4 horas antes de la dosis siguiente para mantener el régimen de dos tomas diarias. No deben tomarse al mismo tiempo las dos dosis. Vómitos: En caso de vómitos tras la administración de vemurafenib, el paciente no debería tomar una dosis adicional del medicamento, y el tratamiento deberá continuarse como de costumbre. Modificación de la dosis (ver Advertencias, Ensayos clínicos y Reacciones adversas): Los acontecimientos adversos sintomáticos o la prolongación del QTc pueden requerir una reducción de la dosis o la retirada temporal o definitiva de Zelboraf. No se recomienda modificar la dosis o interrumpir el tratamiento en el carcinoma cutáneo de células escamosas. No se recomienda reducir la dosis a menos de 480 mg dos veces al día. En caso de que el paciente desarrolle un carcinoma de células escamosas cutáneo (CCEc), se recomienda continuar el tratamiento sin modificar la dosis de vemurafenib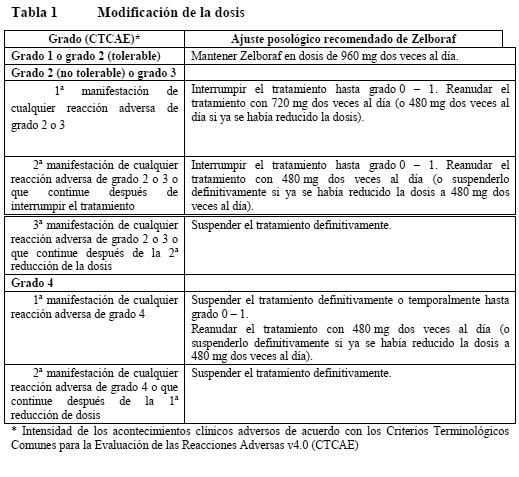
Pautas posológicas especiales: Ancianos: No es necesario ningún ajuste especial de la dosis en los pacientes de 65 o más años. Niños: No se han estudiado la seguridad y la eficacia de Zelboraf en niños y adolescentes ( < 18 años). Insuficiencia renal: No es necesario ajustar la dosis inicial en los pacientes con insuficiencia renal leve o moderada (ver Insuficiencia renal y Farmacocinética en poblaciones especiales). No hay datos suficientes para determinar la potencial necesidad de ajustar la dosis en pacientes con insuficiencia renal grave. No se puede descartar un aumento del riesgo de exposición en pacientes con insuficiencia renal grave. Los pacientes con insuficiencia renal grave deben monitorizarse estrechamente. Insuficiencia hepática: Se dispone de pocos datos en pacientes con insuficiencia hepática. Los pacientes con insuficiencia hepática grave y moderada pueden tener aumentada la exposición y deben monitorizarse estrechamente, ya que el vemurafenib se elimina por el hígado.
Contraindicaciones.
Hipersensibilidad al principio activo o a alguno de los excipientes.
Reacciones adversas.
Ensayos clínicos: Resumen del perfil de seguridad: Las reacciones adversas más comunes (RAM) ( > 30%) notificadas con vemurafenib incluyen artralgia, fatiga, rash, reacciones de fotosensitibilidad, náuseas, alopecia y prurito. Se notificó de manera muy frecuente carcinoma de células escamosas (CCEc) que en la mayoría de los casos fue tratado mediante extirpación local. Listado tabulado de las reacciones adversas: Las reacciones adversas (RAM) que fueron notificadas en los pacientes con melanoma se han enumerado bajo la clasificación de órganos del sistema MedDRA frecuencia y grado de gravedad. La siguiente clasificación se ha utilizado para ordenarlas por frecuencia: Muy frecuentes: ≥ 1/10. Frecuentes: ≥ 1/100 a < 1/10. Poco frecuentes: ≥ 1/1.000 a < 1/100. Raras ≥ 1/10,000 a < 1/1000. Muy raras < 1/10,000. En esta sección, las reacciones adversas a los medicamentos (RAM) se basan en los resultados obtenidos en 468 pacientes de un estudio abierto, aleatorizado, fase III en pacientes adultos con melanoma estadio IV o no resecable con mutación BRAF V600 positiva y también de un estudio fase II, de un solo brazo en pacientes con melanoma estadio IV con mutación BRAF V600 positiva, que habían fallado previamente, al menos a un tratamiento sistémico anterior. Además se notifican las reacciones adversas a los medicamentos (RAMs) procedentes de los informes de seguridad de todos los ensayos clínicos. Todos los términos incluidos están basados en los porcentajes más altos observados entre los ensayos clínicos fases II y III. Dentro de cada categoría por órganos y sistemas, la agrupación de las RAM se presentan en orden decreciente de gravedad y para la evaluación de la toxicidad se notificaron utilizando la escala NCI-CTCAE v 4.0 (Criterios comunes de toxicidad).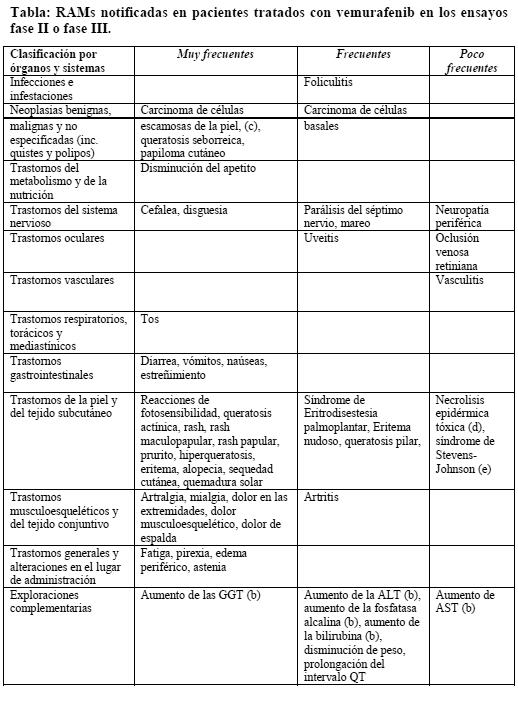
Descripción de las reacciones adversas seleccionadas: Aumento de las enzimas hepáticas (b). Las alteraciones en las enzimas hepáticas notificadas en el ensayo clínico fase III se expresan a continuación como la proporción de pacientes que han experimentado una modificación del valor basal al grado 3 o 4 en las alteraciones de las enzimas hepáticas. Muy frecuentes: GGT. Frecuentes: ALT, fosfatasa alcalina, bilirrubina. Poco frecuentes: AST. La ALT, la fosfatasa alcalina o la bilirrubina, no aumentaron a grado 4. Carcinoma cutáneo de células escamosas (CCCE) (ver Advertencias): La incidencia de CCCE entre los pacientes tratados con vemurafenib en los estudios fue de aproximadamente el 20%. La mayoría de las lesiones excindidas evaluadas por un laboratorio central de dermatología independiente se clasificaron como del subtipo carcinoma de células escamosas-queratoacantoma o con características mixtas de queratoacantoma (52%), que son en ambos casos un tipo más benigno, menos invasivo, de CCCE. La mayoría de las lesiones clasificadas como "otras lesiones" (43%) fueron lesiones cutáneas benignas (p. ej. verruga vulgar, queratosis actínica, queratosis benigna, quiste/quiste benigno). Habitualmente, el CCCE se presentó en una fase temprana del tratamiento: la mediana del tiempo hasta la primera aparición fue de 7 - 8 semanas. De los pacientes con CCCE, aproximadamente el 33% experimentó > 1 episodio, con una mediana del tiempo entre episodios de 6 semanas. El tratamiento habitual consistió en una simple excisión quirúrgica del CCCE y los pacientes pudieron continuar, en general, con el tratamiento sin ajuste de la dosis. Nuevo melanoma primario: En los ensayos clínicos se han notificado casos de nuevos melanomas primarios. Estos casos fueron controlados mediante extirpación, y los pacientes continuaron el tratamiento sin ajustes de dosis. La monitorización de las lesiones cutáneas debe realizarse. Reacciones de hipersensibilidad (ver Advertencias): Se han notificado reacciones graves de hipersensibilidad asociadas con vemurafenib incluyendo anafilaxia. Las reacciones graves de hipersensibilidad pueden incluir el síndrome de Stevens-Johnson, rash generalizado, eritema o hipotensión. En los pacientes que experimenten reacciones graves de hipersensibilidad, se debe interrumpir permanente el tratamiento con vemurafenib. En un estudio clínico (ver resumen de la seguridad) se notificó un caso de reacción de hipersensibilidad con erupción, fiebre, escalofríos e hipotensión a los 8 días de iniciada la administración del vemurafenib en una dosis de 960 mg dos veces al día. Síntomas similares se observaron tras la reanudación del tratamiento con una dosis única de 240 mg de vemurafenib. El paciente suspendió definitivamente la toma de vemurafenib y se recuperó sin secuelas. Reacciones dermatológicas (e): En el ensayo clínico pivotal se han notificado reacciones dermatológicas graves en pacientes que recibieron vemurafenib, incluyendo casos raros del Síndrome de Stevens-Johnson y de necrólisis epidérmica tóxica. En los pacientes que experimenten una reacción dermatológica grave, vemurafenib debe ser interrumpido permanentemente. Prolongación del intervalo QT (ver Advertencias): El análisis de los datos electrocardiográficos centralizados de un subestudio de fase II abierto y no controlado del intervalo QT en 132 pacientes tratados con vemurafenib en una dosis de 960 mg dos veces al día mostró un aumento medio del QTc basal entre el día 1 (3,3 ms; IC superior del 95%: 5 ms) y el día 15 (12,8 ms; IC superior del 95%: 14,9 ms). En este estudio se observó una prolongación del QTc dependiente de la exposición y el efecto medio en el QTc permaneció estable en 12 - 15 ms más allá del primer mes de tratamiento, observándose la prolongación media del QTc más larga (15,1 ms; IC superior del 95%: 17,7 ms) dentro de los 6 primeros meses de tratamiento (n=90 pacientes). En dos pacientes (1,5%) se registraron valores absolutos de QTc relacionados con el tratamiento > 500 ms (grado 3 según los criterios CTCAE), y sólo un paciente (0,8%) presentó un cambio del QTc basal > 60 ms. Los modelos experimentales y la simulación de la prolongación del QT arrojaron las estimaciones siguientes: para la dosis de 960 mg dos veces al día, la predicción del porcentaje de pacientes con una prolongación del QTcP mayor de 60 ms era del 0,05%. Se preveía un aumento del 0,2% de este porcentaje en pacientes obesos con un IMC de 45 kg/m2. El porcentaje previsto de pacientes con un cambio del valor basal de QTcP mayor de 60 ms era del 0,043% en los varones y el 0,046% en las mujeres. El porcentaje de pacientes con valores de QTcP superiores a 500 ms se previó en un 0,05% en los varones y un 1,1% en las mujeres. Alteraciones analíticas: Las alteraciones de las pruebas hepáticas en el estudio clínico de fase III se resumen en la tabla siguiente como la proporción de pacientes que experimentaron un cambio del valor basal al grado 3 o 4.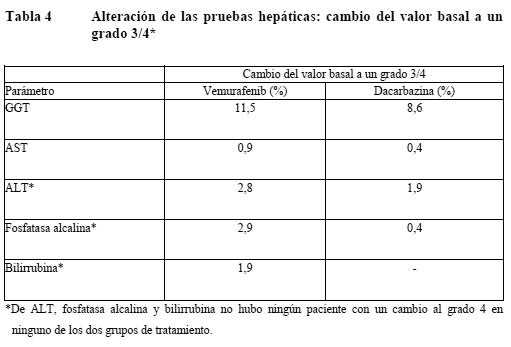
Advertencias.
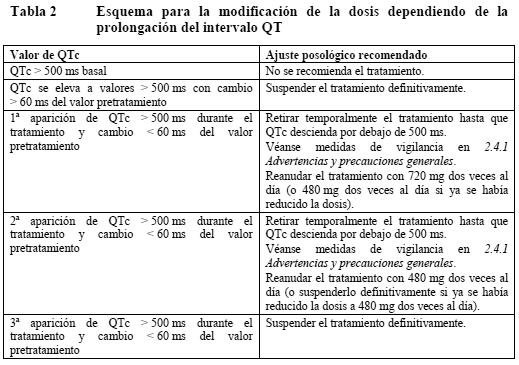
En los pacientes tratados con Zelboraf ha de confirmarse previamente en un test validado la positividad del tumor para la mutación BRAFV600. No se ha establecido de forma convincente, la eficacia y la seguridad de vemurafenib en pacientes con tumores que expresen mutaciones BRAF V600 raras, diferentes a V600E y V600K. Vemurafenib no debería usarse en pacientes con melanoma maligno BRAF de tipo nativo. Carcinoma cutáneo de células escamosas (CCCE): Se han descrito casos de CCCE -incluidos los clasificados como queratoacantoma o subtipo mixto de queratoacantoma- en pacientes tratados con vemurafenib (ver Ensayos clínicos, Reacciones adversas). El CCCE se presentó habitualmente en una fase temprana del tratamiento. Los factores de riesgo potenciales asociados con el CCCE en los estudios clínicos del vemurafenib fueron la edad (≥ 65 años), el cáncer de piel previo y la exposición crónica al sol. El tratamiento habitual consistió en una simple excisión quirúrgica del CCCE y los pacientes pudieron continuar con el tratamiento sin ajustar la dosis. Se recomienda una evaluación dermatológica de todos los pacientes antes de iniciar el tratamiento y controles rutinarios durante el mismo. Toda lesión cutánea sospechosa debe excindirse, enviarse a un análisis dermatológico y tratarse como sea costumbre. La vigilancia debe proseguirse durante 6 meses tras la retirada de Zelboraf o hasta el comienzo de otro tratamiento antineoplásico. Se instará a los pacientes a que informen a su médico si observan algún cambio en la piel. Carcinoma no cutáneo de células escamosas (CnCCE): Se han descrito casos de CnCCE en pacientes que recibían vemurafenib. Antes de iniciarse el tratamiento y cada 3 meses durante el mismo debe realizarse un examen de cabeza y cuello de los pacientes, consistente al menos en un control visual de la mucosa oral y la palpación de ganglios linfáticos. Debe efectuarse, además, una TC torácica antes de empezar el tratamiento y cada 6 meses durante el mismo. Tras la retirada de Zelboraf, la vigilancia de CnCCE debe mantenerse hasta 6 meses o el comienzo de otro tratamiento antineoplásico. Los valores anómalos deben evaluarse como proceda clínicamente. Nuevo melanoma primario: En los ensayos clínicos se han descrito casos de nuevo melanoma primario. Estos casos se controlaron mediante extirpación, y los pacientes continuaron el tratamiento sin ajuste de la dosis. Las lesiones cutáneas deben vigilarse como se indica más arriba en el apartado de CCCE. Reacciones de hipersensibilidad: Se han notificado reacciones de hipersensibilidad graves, anafilaxia inclusive, en asociación con el vemurafenib (ver Ensayos clínicos, Reacciones adversas). Las reacciones de hipersensibilidad graves consistieron en erupción y eritema generalizados o hipotensión. En caso de reacción de hipersensibilidad grave, debe deberá retirarse Zelboraf definitivamente. Reacciones dermatológicas: En el estudio clínico fundamental se han descrito reacciones dermatológicas graves en pacientes tratados con vemurafenib, que en raras ocasiones consistieron en síndrome de Stevens-Johnson y necrólisis epidérmica tóxica. En caso de reacción dermatológica grave, debe retirarse el vemurafenib definitivamente. Prolongación del QT: Se ha observado la prolongación del QT dependiente de la exposición en un subestudio de fase II abierto, no controlado, en pacientes con melanoma metastásico previamente tratados (ver Ensayos clínicos, Reacciones adversas). La prolongación del QT puede elevar el riesgo de arritmias ventriculares, incluida la torsade de pointes (taquicardia ventricular polimorfa en entorchado). No se recomienda el tratamiento con Zelboraf de pacientes con alteraciones electrolíticas (incluyendo al magnesio) no corregibles, síndrome del QT prolongado o que estén tomando medicamentos que prolonguen el intervalo QT. Antes del tratamiento con Zelboraf y después de un mes y tras cada ajuste posológico debe realizarse un control electrocardiográfico y electrolítico (incluyendo al magnesio). Este control debe efectuarse asimismo mensualmente durante los 3 primeros meses de tratamiento; después, cada 3 meses o con mayor frecuencia si se considera indicado clínicamente. No se recomienda empezar el tratamiento con Zelboraf en pacientes con un QTc > 500 ms. Si el QTc sobrepasa los 500 ms (CTCAE ≥ grado 3), se debe retirar temporalmente Zelboraf, corregir las alteraciones electrolíticas y controlar los factores de riesgo cardíaco por prolongación del QT (p. ej.: insuficiencia cardíaca congestiva, bradiarritmias). El tratamiento no debe reiniciarse hasta que el QTc descienda por debajo de 500 ms y debe hacerse con una dosis menor, como se muestra en las Tablas 1 y 2. Se recomienda suspender definitivamente el tratamiento con Zelboraf si, después de corregidos los factores de riesgo asociados, el incremento del QTc es > 500 ms y además existe un cambio > 60 ms respecto del valor pretratamiento. Insuficiencia hepática: No es necesario el ajuste de la dosis inicial para los pacientes con insuficiencia hepática. Los pacientes con insuficiencia hepática moderada debido a metástasis hepática sin hiperbilirrubinemia deben controlarse según las recomendaciones generales. Solo se dispone de datos muy reducidos en pacientes con insuficiencia hepática de moderada a grave. Pacientes con insuficiencia hepática de moderada a grave podrían tener un aumento en su exposición. En consecuencia podría estar justificado realizar un control estrecho, especialmente después de las primeras semanas del tratamiento ya que puede ocurrir una acumulación tras un período largo (varias semanas). Además, se recomienda controlar el ECG de forma rutinaria durante los tres primeros meses. Insuficiencia renal: No es necesario el ajuste de la dosis inicial para los pacientes con insuficiencia renal leve o moderada. Sólo se dispone de datos muy reducidos en pacientes con insuficiencia renal grave. Los pacientes con insuficiencia renal grave deben utilizar vemurafenib con precaución y deben ser controlados estrechamente. Alteraciones de las pruebas hepáticas: Durante el tratamiento con vemurafenib pueden producirse alteraciones de las pruebas hepáticas (ver Alteraciones analíticas, Reacciones adversas). Se deben controlar las enzimas hepáticas (transaminasas y fosfatasa alcalina) y la bilirrubina antes de empezar el tratamiento y mensualmente durante el mismo, o como esté clínicamente indicado. Las medidas ante alteraciones analíticas consistirán en reducir la dosis, interrumpir el tratamiento o suspenderlo definitivamente (ver Dosificación, Modificación de la dosis). Fotosensibilidad: Se ha descrito fotosensibilidad entre moderada a grave en pacientes tratados con vemurafenib en los estudios clínicos (ver Ensayos clínicos, Reacciones adversas). Se aconsejará a todos los pacientes que eviten la exposición solar mientras tomen Zelboraf. Para prevenir quemaduras solares, se aconsejará a los pacientes que, mientras tomen este medicamento, utilicen fuera de casa prendas de vestir protectoras contra el sol, así como un filtro solar UVA-UVB de amplio espectro y protector labial (SPF ≥ 30). En caso de fotosensibilidad de grado 2 (no tolerable) o acontecimientos adversos mayores, se recomienda ajustar la dosis (ver Dosificación, Modificación de la dosis). Reacciones oftalmológicas: Se han notificado reacciones oftalmológicas graves incluyendo uveítis, iritis y oclusión venosa retiniana. Se debe controlar rutinariamente a los pacientes para detectar reacciones oftalmológicas. Efectos sobre la capacidad para conducir y utilizar máquinas: No se han llevado a cabo estudios sobre los efectos del vemurafenib en la capacidad de conducir vehículos y utilizar maquinaria. Pruebas de laboratorio: ver Advertencias.
Interacciones.
Efectos de vemurafenib en los sustratos CYP: Cuando se administró de forma conjunta una dosis única de cafeína después de la administración de dosis repetidas de vemurafenib durante 15 días se observó inhibición del CYP1A2. Esto dio como resultado un incremento medio de 2,5 veces (el máximo fue de 10 veces) de los niveles plasmáticos de cafeína tras el tratamiento con vemurafenib. Vemurafenib puede incrementar los niveles plasmáticos de sustancias que se metabolizan predominantemente por CYP1A2 y se debe considerar un ajuste de dosis. Cuando se administró de forma conjunta una dosis única de midazolam tras la administración repetida de vemurafenib durante 15 días se observó una inducción del CYP3A4. Esto dio como resultado una disminución media del 32% (con un máximo de hasta un 80%) de los niveles plasmáticos de midazolam tras el tratamiento con vemurafenib. Vemurafenib puede disminuir los niveles plásmaticos de sustancias que se metabolizan predominantemente por CYP3A4. Basándonos en esto, la eficacia de los anticonceptivos orales que se metabolizan vía CYP3A4 y que se utilicen de forma concomitante con vemurafenib podría verse disminuida. Se debe considerar un ajuste de dosis para los sustratos de CYP3A4 con estrecha ventana terapéutica. Se ha observado in vitro una inducción leve del CYP2B6 por vemurafenib a una concentración de vemurafenib de 10 mM. Se desconoce actualmente si vemurafenib a niveles de plasmáticos de 100 mM observados en los pacientes en estado estacionario (aproximadamente 50 mg/ml) podría disminuir las concentraciones plasmáticas de sustratos CYP2B6 administrados concomitantemente, tales como bupropión. Algunos pacientes mostraron un aumento en los niveles de warfarina (una media de un 20%) cuando se administró de forma conjunta una dosis única de warfarina tras la administración repetida de vemurafenib durante 15 días. Debe tenerse precaución cuando se administra de forma conjunta vemurafenib y warfarina (CYP2C9) en pacientes con melanoma. Vemurafenib inhibió CYP2C8 in vitro. Se desconoce la relevancia in vivo de este hallazgo, pero no se puede excluir un riesgo de un efecto clínicamente relevante sobre la administración concomitante de sustratos del CYP2C8. Debido a la larga semivida de vemurafenib, el efecto inhibitorio completo de vemurafenib sobre un medicamento concomitante podría no ser observado antes de los ocho primeros días de tratamiento con vemurafenib. Tras la finalización del tratamiento con vemurafenib, puede ser necesario un período de lavado de 8 días para evitar interacciones con el tratamiento posterior. Efectos de vemurafenib en los sistemas transportadores de medicamentos: Estudios in vitro han demostrado que vemurafenib es un inhibidor del transportador (P-gp). Se desconoce la relevancia clínica de esta información. No se puede excluir que vemurafenib pueda aumentar los niveles plasmáticos de otros medicamentos transportados por P-gp. Actualmente se desconoce el posible efecto de vemurafenib en otros transportadores (ej., BCRP). Efectos de la administración concomitante de otros medicamentos con vemurafenib Estudios in vitro sugieren que el metabolismo de CYP3A4 y la glucuronidación son responsables del metabolismo de vemurafenib. Parece ser que la excreción biliar es otra importante vía de eliminación. No existen datos clínicos disponibles que muestren la actividad de inducción o inhibición fuerte del CYP3A4 y/o la actividad de las proteínas trasportadoras sobre los niveles de vemurafenib. Vemurafenib se debe utilizar con precaución en combinación con inhibidores potentes del CYP3A4, de la glucoronidación, y/o de las proteínas trasportadoras (ej., ritonavir, saquinavir, telitromicina, ketoconazol, itraconazol, voriconazol, posaconazol, nefazodona, atazanavir). La administración concomitante de inductores potentes del P-gp, de la glucuronidación, y/o del CYP3A4 (ej. rifampicina, rifabutina, carbamazepina, fenitoína o hierba se San Juan [Hypericum perforatum]) puede dar lugar a niveles subóptimos de vemurafenib y debería ser evitada. Estudios in vitro han demostrado que vemurafenib es un sustrato del trasportador P-gp. Se desconocen los efectos de los inductores e inhibidores del P-gp sobre los niveles de vemurafenib. No se puede excluir que la farmacocinética de vemurafenib pueda verse afectada por los medicamentos que inhiben o influyen sobre P-gp (ej. verapamilo, claritromicina, ciclosporina, ritonavir, quinidina, dronedarona, amiodarona,