ZEMPLAR®
ABBOTT
Análogo sintético de la vitamina D.
Descripción.
Paricalcitol, USP, el ingrediente activo en el paricalcitol inyectable, es un análogo de calcitriol manufacturado sintéticamente, la forma metabólicamente activa de la vitamina D. Paricalcitol inyectable está disponible como una solución acuosa, incolora, clara y estéril para inyección intravenosa. Cada 1 mL contiene Paricalcitol 5 mcg; propilen glicol, 30% (v/v) y alcohol (etanol) 20% (v/v.). Paricalcitol es un polvo blanco, cristalino con la fórmula empírica de C27H44O3, que corresponde a un peso molecular de 416.64. Paricalcitol está químicamente designado como 19-nor-1a, 3b, 25-trihidroxi-9,10- secoergosta-5(Z), 7(E), 22(E)-triene.
Farmacología.
Propiedades Farmacodinámicas: El hiperparatiroidismo secundario se caracteriza por una elevación en la hormona paratiroidea (PTH) asociado con niveles inadecuados de la hormona vitamina D activa. La fuente de vitamina D en el cuerpo es desde la síntesis en la piel y desde la ingesta alimentaria. La vitamina D requiere dos hidroxilaciones secuenciales en el hígado y el riñón para unirse y activar al receptor de vitamina D (RVD). El activador del RVD endógeno, calcitriol [1,25(OH)2D3], es una hormona que se une a los RVDs que están presentes en la glándula paratiroidea, intestino, riñón y huesos para mantener la función paratiroidea y la homeostasis del calcio y fósforo, y se une a los RVDs encontrados en muchos otros tejidos, incluyendo próstata, endotelio y células inmunes. La activación del RVD es esencial para la apropiada formación y mantenimiento del hueso normal. En el riñón enfermo, la activación de vitamina D está disminuida, produciendo un aumento de PTH, llevando subsecuentemente a un hiperparatiroidismo secundario, y trastornos en la homeostasis del calcio y fósforo1. Los niveles disminuidos de 1,25(OH)2D3 y la resultante de niveles elevados de PTH, los cuales a menudo preceden las anormalidades en el calcio y fósforo sérico, afectan la tasa de recambio óseo y pueden producir osteodistrofia renal. Las reducciones en PTH en los pacientes con enfermedad renal crónica (ERC) se asocian con un impacto favorable sobre las fosfatasas alcalinas específicas del hueso, recambio óseo y fibrosis ósea. Además de reducir la PTH y corregir el recambio óseo, la terapia con vitamina D activa puede prevenir o tratar otras consecuencias de la deficiencia de vitamina D. Mecanismo de Acción: El paricalcitol es un análogo de vitamina D del calcitriol, sintético, biológicamente activo con modificaciones en la cadena lateral (D2) y el anillo A (19-nor). Los estudios preclínicos e in vitro han demostrado que las acciones biológicas del paricalcitol son mediadas a través de la unión al RVD, lo que produce la activación selectiva de las vías de respuesta de la vitamina D. La vitamina D y el paricalcitol han mostrado que reducen los niveles de hormona paratiroidea inhibiendo la síntesis y secreción de PTH. Los niveles disminuidos de 1,25(OH)2D3 se han observado en etapas tempranas de enfermedad renal crónica (ERC). Propiedades Farmacocinéticas: Dentro de dos horas después de la administración de dosis en un rango desde 0.04 a 0.24 mcg/kg, las concentraciones de paricalcitol disminuyeron rápidamente; por lo tanto, las concentraciones de paricalcitol declinaron log-linealmente con una media de vida media cercana a 15 horas. No se observó acumulación de paricalcitol con múltiples dosis. Distribución: Paricalcitol se une extensamente a las proteínas plasmáticas ( > 99%). En sujetos sanos, el volumen de distribución en estado estable es aproximadamente 23.8 L. El volumen medio aparente de distribución después de una dosis de 0.24 mcg/kg de paricalcitol en sujetos con ERC etapa 5 que requieren hemodiálisis (HD) y peritoneodiálisis (PD) es entre 31 y 35 L. La farmacocinética de paricalcitol se ha estudiado en pacientes con insuficiencia renal crónica ERC Etapa 5 que requirieron hemodiálisis. El paricalcitol se administró como una inyección intravenosa en bolo. Metabolismo: Varios metabolitos se detectaron tanto en la orina como en las heces, sin paricalcitol detectable en la orina. Los datos in vitro sugieren que el paricalcitol se metaboliza por múltiples enzimas hepáticas y no-hepáticas, incluyendo CYP24 mitocondrial, así como CYP3A4 y UGT1A4. Los metabolitos identificados incluyen el producto de 24(R)-hidroxilación (presente en bajos niveles en el plasma), también el 24,26- y 24,28-dihidroxilación y glucuronidación directa. Paricalcitol no es un inhibidor de CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 o CYP3A en concentraciones hasta 50nM (21 ng/mL). Se observó una inducción menos del doble para CYP2B6, CYP2C9 y CYP3A4 en concentraciones similares de paricalcitol. Eliminación: El paricalcitol se elimina principalmente por excreción hepatobiliar. Aproximadamente 63% de la radioactividad se eliminó en las heces y 19% se recuperó en la orina en sujetos sanos. En sujetos sanos, la media de la vida media de eliminación de paricalcitol es cerca de cinco a siete horas en el rango de dosis estudiadas de 0.04 a 0.16 mcg/kg.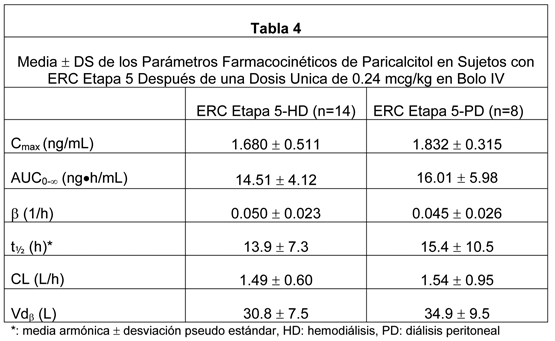
Poblaciones Especiales: Geriátricos: La farmacocinética de paricalcitol no se ha investigado en pacientes geriátricos mayores de 65 años. Pediátricos: La farmacocinética de paricalcitol no se ha investigado en pacientes pediátricos menores de 18 años de edad. Género: La farmacocinética de paricalcitol fue independiente del género. Alteración hepática: La disposición de paricalcitol (0.24 mcg/kg) se comparó en pacientes con alteración hepática leve (n=5) y moderada (n=5) (según lo indicado por el método Child-Pugh) y sujetos con función hepática normal (n=10), La farmacocinética del paricalcitol no unido fue similar a través del rango de función hepática evaluada en este estudio. No se requiere ajuste de dosis en pacientes con alteración hepática leve y moderada. La influencia de la alteración hepática severa sobre la farmacocinética de paricalcitol no se ha evaluado. Alteración renal: La farmacocinética de paricalcitol se ha estudiado en sujetos con ERC Etapa 5 que requirieron hemodiálisis (HD) y diálisis peritoneal (PD). El procedimiento de hemodiálisis no tiene efecto esencialmente sobre la eliminación de paricalcitol. Sin embargo, comparado a sujetos sanos, los sujetos con ERC Etapa 5 mostraron un CL disminuido y una vida media aumentada. Interacciones de Drogas: Un estudio in vitro indica que el paricalcitol no es un inhibidor de CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 o CYP3A en concentraciones hasta 50nM (21 ng/mL) (aproximadamente 20 veces mayor que lo obtenido después de la dosis más alta testeada). En cultivos frescos primarios de hepatocitos, la inducción observada en las concentraciones de paricalcitol de hasta 50 nM fue menos de dos veces para CYP2B6, CYP2C9 o CYP3A, donde los controles positivos presentaron una inducción de seis a diecinueve veces. Por lo tanto, no se espera que paricalcitol inhiba o induzca el clearance de drogas metabolizadas por estas enzimas. No se han estudiado interacciones de drogas con paricalcitol inyectable. Ketoconazol: Aunque no se estudió con paricalcitol inyectable, el efecto de múltiples dosis de ketoconazol administrados como 200 mg dos veces al día por 5 días sobre la farmacocinética de paricalcitol cápsulas se ha estudiado en sujetos sanos. El Cmáx de paricalcitol se afectó mínimamente, pero el AUC0-inf aproximadamente se dobló en presencia de ketoconazol. La media de la vida media de paricalcitol fue 17.0 horas en presencia de ketoconazol comparado con 9.8 horas, cuando el paricalcitol se administró solo (ver Advertencias). Datos de seguridad pre-clínicos: Carcinogénesis: En un estudio de carcinogenicidad de 104 semanas en ratones CD-1, se observó un aumento de la incidencia de leiomioma uterino y de leiomiosarcoma con dosis subcutáneas de 1, 3, 10 mcg/kg (2 a 15 veces el AUC de la dosis humana de 14 mcg, equivalente a 0.24 mcg/kg basado en AUC). La tasa de incidencia de leiomioma uterino fue significativamente diferente que el grupo control en la dosis más alta de 10 mcg/kg. En un estudio de carcinogenicidad en 104 semanas en ratas, había una incidencia creciente de feocromocitoma suprarrenal benigno en dosis subcutáneas de 0,15 a 1,5 mcg/kg ≤1 veces el máximo recomendado para humanos, dosis semanal de 0,72 mcg/kg, basados en el área superficial corporal, mg/m2). En un estudio de carcinogenicidad de 104 semanas en ratas, hubo un aumento de la incidencia de feocromocitoma adrenal benigno con dosis subcutáneas de 0.15, 0.5, 1.5 mcg/kg ( < 1 a 7 veces la exposición después de una dosis humana de 14 mcg, equivalente a 0.24 mcg/kg basado en AUC). El aumento de la incidencia de feocromocitomas en ratas se puede relacionar con la alteración de la homeostasis del calcio por paricalcitol. Mutagénesis: El paricalcitol no exhibió toxicidad genética in vitro con o sin activación metabólica en el ensayo de mutagénesis microbiana (Ensayo de Ames), ensayo de mutagénesis de linfoma en ratones (L5178Y), o un ensayo de aberración cromosómica en células linfocito humano. Tampoco hubo evidencia de toxicidad genética en un ensayo de micronúcleos de ratón in vivo. Deterioro de la Fertilidad: El paricalcitol no tuvo efecto sobre la fertilidad (macho o hembra) en ratas con dosis intravenosas de hasta 20 mcg/kg/dosis [equivalente a 13 veces la dosis humana de 14 mcg (0.24 mcg/kg) basado en área de superficie, mcg/m2]. Estudios clínicos: Pacientes adultos: Los estudios en pacientes con insuficiencia renal crónica (ERC etapa 5) muestran que paricalcitol suprime los niveles de PTH sin una diferencia significativa en la incidencia de hipercalcemia o hiperfosfatemia cuando se comparó con placebo. Sin embargo, el fósforo, calcio y producto calcio x fósforo (Ca x P) sérico pueden aumentar cuando se administra paricalcitol. En tres estudios fase III, de 12 semanas, placebo controlados en pacientes con insuficiencia renal crónica (ERC etapa 5) en diálisis, la dosis de paricalcitol se comenzó en 0,04 mcg/kg tres veces por semana. La dosis se aumentó en 0,04 mcg/kg cada dos semanas hasta que los niveles de hormona paratiroidea intacta (iPTH) se disminuyeron al menos 30% desde el estado basal o en una quinta escalada se llevó la dosis a 0,24 mcg/kg, o la iPTH cae a menos de 100 pg/mL, o el producto Ca x P fue mayor que 75 dentro de cualquier período de dos semanas, o el calcio sérico se volvió mayor que 11,5 mg/dL en cualquier momento. Los pacientes tratados con paricalcitol alcanzaron una reducción media de iPTH de 30% dentro de seis semanas. En estos estudios, no hubo diferencia significativa en la incidencia de hipercalcemia o hiperfosfatemia entre los pacientes tratados con paricalcitol y con placebo. Los resultados de estos estudios son los siguientes: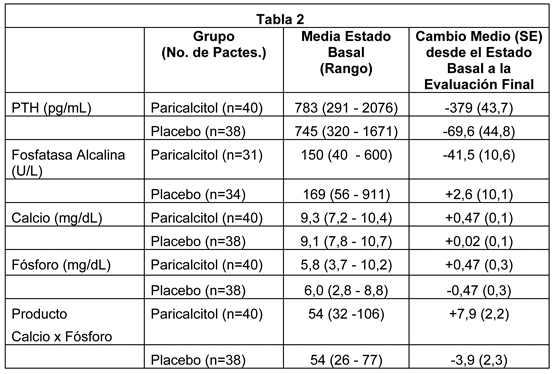
En un estudio fase 4, doble-ciego, randomizado, multicéntrico, 12 semanas, paricalcitol se administró en una dosis inicial de ya sea 0.04 mcg/kg o tres veces la iPTH basal/80 semanalmente a pacientes con insuficiencia renal crónica (ERC etapa 5) en diálisis. La dosis se incrementó en 2 mcg cada dos semanas hasta que los niveles de PTH descendieran entre 30% a 60% desde el estado basal, o la iPTH descendiera a menos de 100 pg/mL, o el producto Ca x P fuera mayor que 75 en dos ocurrencias consecutivas, o el calcio sérico empieza a ser mayor que 11.5 mg/dL en cualquier momento. Los pacientes completaron el estudio reduciendo la iPTH ≥30% desde los niveles basales en cuatro mediciones consecutivas, o teniendo una incidencia única de hipercalcemia, o completando doce semanas de tratamiento. No se observaron incidencias de hipercalcemia en ningún paciente en ningún grupo de tratamiento. Ambos métodos de dosificación mostraron ser seguros y efectivos. Los resultados se presentan en la siguiente tabla.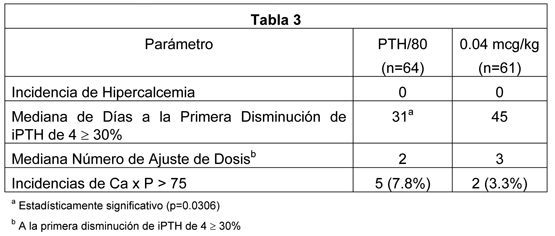
Un estudio de seguridad abierto, a largo plazo en 164 pacientes con ERC etapa 5 (dosis media de 7,5 mcg tres veces por semana), demostró que la media del Ca, P y Ca x P séricos permanecieron dentro de los rangos clínicamente apropiados con reducción de la PTH (disminución media de 319 pg/mL en 13 meses.)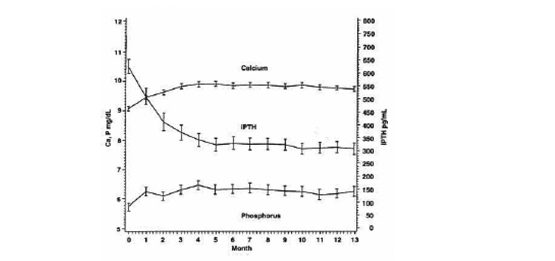
Pacientes Pediátricos: Se analizó la seguridad y la efectividad de paricalcitol inyectable en un estudio de 12 semanas, randomizado, doble ciego, placebo-controlado en 29 pacientes pediátricos, de 5-19 años de edad, con enfermedad renal en etapa terminal en hemodiálisis. Casi todos los pacientes habían recibido alguna forma de vitamina D previo al estudio. Setenta y seis por ciento de los pacientes eran masculinos, 52% eran caucásicos y 45% eran Africano-Americanos. La dosis inicial de paricalcitol fue de 0.04 mcg/kg 3 veces a la semana, basada en el nivel de iPTH basal de < 500 pg/mL, o 0.08 mcg/kg 3 veces a la semana basado en el nivel de iPTH basal de ≥500 pg/mL, respectivamente. La dosis de Paricalcitol se ajustó en incrementos de 0.04 mcg/kg basado en los niveles séricos de iPTH, calcio, y Ca x P. Los niveles medios basales de iPTH fueron de 841 pg/mL para los 15 pacientes tratados con paricalcitol y 740 pg/mL para los 14 pacientes tratados con placebo. La dosis media de paricalcitol administrada fue de 4.6 mcg (rango: 0.8 mcg - 9.6 mcg). Diez de los 15 pacientes (67%) tratados con paricalcitol y 2 de los 14 pacientes (14%) tratados con placebo completaron el estudio. Diez de los pacientes tratados con placebo (71%) se discontinuaron debido a elevaciones excesivas en los niveles de iPTH definida por 2 mediciones consecutivas de niveles de iPTH > 700pg/mL y mayor que el estado basal después de 4 semanas de tratamiento. En el análisis de eficacia primaria, 9 de 15 pacientes (60%) en el grupo paricalcitol tenían 2 disminuciones consecutivas de 30% desde el estado basal de iPTH comparado con 3 de 14 pacientes (21%) en el grupo placebo (95% CI para la diferencia entre grupos -1%, 63%). Durante el estudio, ningún paciente ya sea en el grupo paricalcitol o grupo placebo desarrollaron hipercalcemia (definida como al menos un valor de calcio > 11.2 mg/dL).
Indicaciones.
Paricalcitol inyectable está indicado para la prevención y tratamiento del hiperparatiroidismo secundario asociado con insuficiencia renal crónica.
Dosificación.
La vía usual de administración de la solución de paricalcitol para inyección es vía conector sanguíneo a través del acceso de hemodiálisis. Para pacientes sin acceso de hemodiálisis, las inyecciones de paricalcitol se deben administrar a través de inyección intravenosa lenta, por no menos de 30 segundos, para minimizar el dolor en la administración. Adultos: Dosis Inicial: Existen dos métodos alternativos para determinar la dosis inicial de paricalcitol. La dosis máxima administrada con seguridad en estudios clínicos fue tan alta como 40 microgramos. Dosis Inicial Basada en Peso Corporal: La dosis inicial recomendada de paricalcitol es 0.04 mcg/kg a 0.1 mcg/kg (2.8 - 7 mcg) administrado como una dosis en bolo no más frecuentemente que día por medio en cualquier momento durante la diálisis. Dosis Inicial Basada en Niveles de iPTH basales: Una segunda generación de ensayo de PTH (PTH intacta [iPTH]) se ha usado como medida de la PTH biológicamente activa en pacientes con insuficiencia renal crónica (ERC etapa 5). La dosis inicial se calcula por la siguiente fórmula y se administra como una dosis en bolo intravenoso (IV) no más frecuentemente que día por medio en cualquier momento durante la diálisis:
Dosis de Titulación: El rango objetivo aceptado actualmente para niveles de PTH en la etapa terminal de los pacientes con enfermedad renal que se encuentran en diálisis no es más de 1,5 a 3 veces el límite superior no urémico del normal (150-300 pg/mL para iPTH). La supervisión cercana y la titulación individual de la dosis son necesarias para alcanzar los resultados fisiológicos apropiados. Durante cualquier período de ajuste de dosis, el calcio sérico (corregido para hipoalbuminemia) y los niveles de fósforo se deben monitorear más frecuentemente. Si se observan un nivel corregido elevado de calcio (Ca) ( > 11,2 mg/dL) o niveles persistentemente elevados de fósforo (P) ( > 6.5 mg/dL), la dosificación de la droga se debe ajustar hasta que estos parámetros se normalicen. Si se observa hipercalcemia o un producto Ca x P corregido persistentemente elevado, mayor que 75, la dosificación de la droga se debe reducir o interrumpir hasta que estos parámetros se normalicen. Entonces, la administración de paricalcitol se debe reiniciar en una menor dosis. Si un paciente se encuentra con un quelante de fósforo basado en calcio, la dosis se puede disminuir o discontinuar, o el paciente se puede cambiar a un quelante de fósforo no basado en calcio. Las dosis pueden necesitar ser disminuidas por la disminución de los niveles de PTH en respuesta a la terapia. Así, la dosis incremental se debe individualizar. Si no se observa una respuesta satisfactoria, la dosis se puede aumentar en 2 a 4 mcg en intervalos de 2 a 4 semanas. Si en cualquier momento el nivel de iPTH disminuye a menos de 150 pg/mL, la dosis de la droga se debe disminuir. La siguiente tabla es una aproximación sugerida para la titulación de la dosis: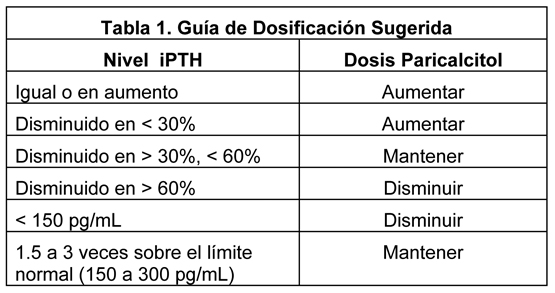
Los productos de drogas parenterales se deben inspeccionar visualmente por material partículado y decoloración previo a la administración cada vez que la solución y el contenedor lo permitan. Eliminar la porción no empleada.
Contraindicaciones.
Paricalcitol inyectable no se debe administrar a pacientes con evidencia de toxicidad por vitamina D, hipercalcemia o hipersensibilidad a cualquier componente de este producto (ver Advertencias.)
Reacciones adversas.
Eventos Adversos de los Estudios Clínicos Fase 2 y Fase 3: En cuatro estudios multicéntricos, doble ciego, placebo-controlados la discontinuación de la terapia debido a cualquier evento adverso ocurrió en 6,5% de 62 pacientes tratados con paricalcitol (dosis tituladas como toleradas, ver Farmacología, Estudios clínicos) y 2,0% de 51 pacientes tratados con placebo por uno a tres meses. Los eventos adversos que ocurrieron con mayor frecuencia en el grupo paricalcitol con una frecuencia de 2% o más, sin relación de causalidad, se presentan en la siguiente tabla: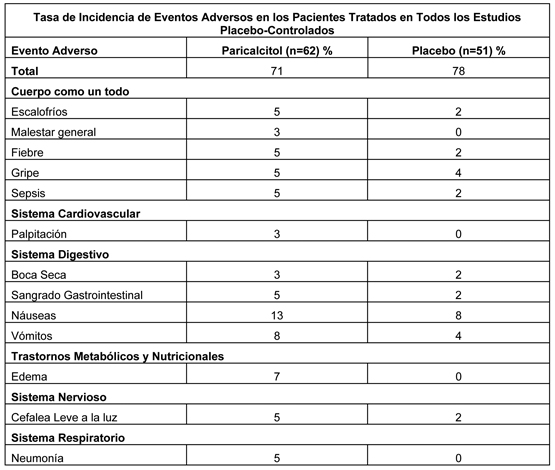
Un paciente que informó el mismo término médico más de una vez se contabilizó sólo una vez para ese término médico. Los parámetros de seguridad (cambios en la media de Ca, P, Ca x P) en un estudio de seguridad abierto hasta trece meses de duración apoyan la seguridad a largo plazo de paricalcitol en esta población de pacientes. Eventos Adversos de Estudios Clínicos Fase 4: En un estudio fase 4 de Hallazgo de Dosis se reportaron comúnmente cefalea (2%) y perversión del gusto (2%) (ver Estudios clínicos para una descripción del estudio). Reacciones Adversas de la Experiencia Post-Comercialización: Las siguientes reacciones adversas se han reportado en la experiencia post-comercialización con paricalcitol inyectable. Las reacciones adversas se presentan por sistema órgano clase. Trastornos del sistema inmune: reacción alérgica, urticaria, angioedema y edema laríngeo. Trastornos del sistema nervioso: perversión del gusto (sabor metálico). Trastornos de la piel y tejido subcutáneo: rash, prurito.
Advertencias.
La sobredosis aguda de paricalcitol puede causar hipercalcemia y requiere atención de emergencia. Durante el ajuste de dosis, se deben monitorear los niveles de calcio y fósforo sérico rigurosamente. Si se desarrolla hipercalcemia clínicamente significativa, la dosis se debe reducir o interrumpir. La administración crónica de paricalcitol puede colocar a los pacientes en riesgo de hipercalcemia, producto Ca x P elevado y calcificación metastásica. El tratamiento de pacientes con hipercalcemia clínicamente significativa consiste en la reducción inmediata de la dosis o interrupción de la terapia con paricalcitol e incluye una dieta con bajo contenido de calcio, e interrupción de los suplementos de calcio, movilización del paciente, atención a los desequilibrios de líquidos y electrolitos, evaluación de anormalidades electrocardiográficas (crítico en pacientes que reciben digitálicos) y hemodiálisis o diálisis peritoneal contra un dializado libre de calcio, para seguridad. Los niveles séricos de calcio se deben monitorear frecuentemente hasta alcanzar la normocalcemia. Los componentes de fosfato o vitamina D-relacionados no se deben tomar concomitantemente con paricalcitol. La toxicidad a digitálicos se potencia por hipercalcemia de cualquier causa, por lo tanto se debe tener precaución cuando los compuestos digitálicos se prescriben junto con paricalcitol. Si los niveles de PTH se suprimen a niveles anormales, se pueden desarrollar lesiones óseas adinámicas (enfermedad ósea de bajo recambio). Pruebas de Laboratorio: Durante el ajuste de dosis y antes de que se establezca la dosis con paricalcitol, los tests de laboratorio se pueden requerir más frecuentemente. Una vez que la dosis se ha establecido, el fósforo y el calcio séricos se deben medir al menos mensualmente. Se recomiendan mediciones de PTH sérica o plasmática cada tres meses (ver Dosis y Administración). Se recomienda una segunda generación o pruebas más tardías de PTH para una detección confiable de la PTH biológicamente activa en pacientes con ERC Etapa 5.
Interacciones.
No se espera que paricalcitol inhiba el clearance de drogas metabolizadas por enzimas del citocromo P450 CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 o CYP3A ni que induzca el clearance de droga metabolizada por CYP2B6, CYP2C9 o CYP3A. No se han realizado estudios específicos de interacción con paricalcitol inyectable. Un estudio de interacción droga-droga dosis múltiple con ketoconazol y paricalcitol cápsula demostró que el ketoconazol aproximadamente dobló el AUC0-infde paricalcitol (ver Farmacología - Interacciones de Droga). Como el paricalcitol se metaboliza parcialmente por el CYP3A y se sabe que el ketoconazol es un potente inhibidor de la enzima 3A del citocromo P450, se debe tener precaución cuando se dosifica paricalcitol con ketoconazol y otros inhibidores potentes de P450 3A. No deben administrarse en toma prolongada preparados que contengan aluminio (ej. antiácidos, ligantes de fosfato), con derivados de Vitamina D, ya que pueden aumentar los niveles sanguíneos de aluminio y puede producirse toxicidad por aluminio a nivel óseo. Altas dosis de preparados que contengan calcio o diuréticos tiazídicos pueden aumentar el riesgo de hipercalcemia. No se deben administrar conjuntamente preparados que contengan magnesio en conjunto con preparados de vitamina D, ya que puede ocurrir hipermagnesemia. Embarazo: Se ha mostrado que el paricalcitol causa disminuciones mínimas en la viabilidad fetal (5%) cuando se administra diariamente a conejos en una dosis de 0,5 veces la dosis humana de 0,24 mcg/kg (basado en área de superficie, mcg/m2) y cuando se administra en ratas en una dosis dos veces la dosis humana de 0,24 mcg/kg (basado en niveles plasmáticos de exposición). En la dosis más alta testeada (20 mcg/kg tres veces por semana en ratas, 13 veces la dosis humana de 0,24 mcg/kg basada en área de superficie), hubo un significativo aumento de la mortalidad de ratas recién nacidas en dosis que fueron maternalmente tóxicas (hipercalcemia). No se observaron otros efectos en el desarrollo de la descendencia. El Paricalcitol no fue teratogénico en las dosis testeadas. No existen estudios adecuados y bien controlados en mujeres embarazadas. El paricalcitol se debe usar durante el embarazo sólo si el beneficio potencial para la madre justifica el riesgo potencial al feto. Madres en Lactancia: Estudios en ratas han mostrado que el paricalcitol está presente en la leche. No se sabe si el paricalcitol se excreta en la leche humana. En la paciente que amamanta, se debe tomar la decisión de si discontinuar la lactancia o discontinuar la droga, tomando en consideración la importancia de la droga para la madre. Uso Pediátrico: Existe experiencia limitada con el uso de paricalcitol inyectable en pacientes menores de 18 años de edad (ver Estudios clínicos - Pacientes Pediátricos). Uso Geriátrico: De los 40 pacientes que recibieron paricalcitol en los tres estudios de Insuficiencia Renal Crónica, placebo-controlados, fase III, diez pacientes tenían 65 años o más. En estos estudios, en general no se observaron diferencias en eficacia o seguridad entre pacientes de 65 años o mayores y pacientes más jóvenes.
Conservación.
Almacenar a 25°C (77°F). Variaciones permitidas de 15 a 30°C (59 - 86°F).
Sobredosificación.
La sobredosis de paricalcitol puede llevar a una hipercalcemia (ver Advertencias.)
Presentación.
Paricalcitol Inyectable de 5 mcg/mL se presenta en Frasco ampolla de dosis única de 1 y 2 mL.